2022年4月14日,美国国家癌症研究所 (NCI) 计算机辅助药物设计研究课题组的Wendy等人在JCIM杂志上发表综述,整理了当前用于药物研发的超大型数据库以及检索技术的情况。

摘要
更廉价、更快速地设计新药与更广泛、更高效地探索化学空间密切相关。可探索的化学空间是巨大的,最近计算机软件和硬件的进步使研究人员能够探索包含数十亿个化学结构的虚拟化学空间。
本文介绍了许多包含百万甚至数十亿化学结构的数据集,以及未完全枚举的更大的化学空间。我们给出了化学库和空间的案例以及用来构造它们的手段,讨论了在化学空间中搜索大型库和组合搜索的新技术。
我们还介绍了空间定位技术,并考虑了设计新药的新方法以及“自主实验室”对设计化合物合成的影响。最后,我们总结了未来的一些其他挑战和机遇。
前言
为了更廉价、更快速地生产新药,研究人员需要使药物发现的“设计-合成-试验-分析 (DMTA) ”周期更有效率。这样做的方法包括更好地探索化学空间,制造更多的化合物 (最好具有药物性质),以及找到更廉价和更有效地合成或购买化合物的方法。化学空间是巨大的,但最近计算机软件和硬件的进步使研究人员能够更广泛或更仔细地探索它。研究人员现在可以定位包含数十亿个化学结构的虚拟化学空间 (图1),进行相似搜索和虚拟筛选,研究结构-活性关系,进行骨架跃迁实验和其他药物发现等方法。

图1.目前已经建立的超大型化合物数据集
商业库 (大写字母、方块)、商业DNA编码库 (大写字母,双三角形)、专有空间 (数字,钻石) 和公共合集 (小写字母、球体)。颜色是对化合物合成可及性的预测,分为100% (蓝色)、非常高 (绿色)、高 (橙色) 和中低 (红色)。
本文介绍了数百万甚至数十亿枚举化学结构的集合,以及通过未完全枚举的大型化学空间进行直接搜索的方法。我们把‘空间’与‘库’、‘库’与‘数据库’区分开来。空间是组合构造的化合物集合,它们通常的确很大,很难列举所有被覆盖的精确化学结构;化合物库是列举式的全结构的集合,通常少于109个分子;数据库则是将化合物库存储到关系数据库管理系统中。
化合物库与空间示例
雷蒙德的团队列举了按照化学稳定性和合成可行性的简单规则假设可以制造的分子,形成生成数据库 (Generated DataBases, GDB) :GDB-11、GDB-13和GDB-17。GDB-17含有166.4亿个由C、N、O、S原子和卤素分子等17个原子构成的分子。GDB的子集和变体也已建成。关于GDB的工作是开创性的:首次尝试完全描述小有机化合物的化学空间。然而,对于实际应用,人们不得不记住,这些分子的大部分从未被合成,甚至合成途径也大多未知。
雷蒙德等人估计GDB-18比GDB-17大50倍,因此他们与阿斯利康合作研究了抽样与穷举计数。他们对用不同SMILES变体的不同大小GDB-13子集训练的模型进行了基准测试,模型有两种不同的循环单元类型,以及不同的超参数组合。开发了新的度量指标,定义了生成的化学空间的均匀性、封闭性和完备性。结果表明,使用100万随机SMILES训练的长短时记忆细胞的模型比同类方法能够产生更大的化学空间,更准确地表示目标化学空间。
合成可获得性虚拟库 (Synthetically Accessible Virtual Inventory, SAVI) 背后的概念不是设计一个后来被证明是不可合成的分子,而是生产一个能够可靠、廉价合成的化合物集合。SAVI是由一组可靠可用且价格低廉的起始材料产生的,其使用的是高度预测且注释丰富的转换规则,该项目是用最早应用于合成分析的逻辑和启发式 (LHASA) 的CHMTRN/PATRAN语言对编写的。有些转换取自原LHASA知识库,但大多是为SAVI项目新编写的。所用的起始原料最初来自Millipore Sigma,但后来被Enamine的构筑单元取代为SAVI-2020。通过LHASA类型转换连接构建块的化学信息学引擎是CACTVS。
虽然CHMTRN是定义化学合成知识的有力工具,但它是一种旧的、非结构化的、非标准化的语言,在前向合成的语境中呈现出挑战。为了解决这些问题,SAVI团队正在开发SMARTS和基于逻辑协助的SLICE项目。SLICE旨在允许化学家轻松地对化学知识进行编码,并通过直接在一个分子上进行推理来完成SMARTS。它将集成在一个平台中,使没有编程语言经验的用户能够以图形化的方式对化学知识进行编码,并将在生成产品的构筑单元上快速执行SLICE支持的转换。
许多化学样品的商业供应商提供由高度稳定的化学反应构建的大型“按需制造”系列和可用的构建块。这些是图1中标明"商业"的化合物库或空间;它们含有具有很高商业可用性的化学物质,任何人都可以寻找它们。它们通常是可以合成的,因为它们的结构单元是可购买的。另外,平行合成方法是稳健的,并且根据类似的先前反应的数据提前预测了合成的可行性。
ZINC是一个公共可用的化合物库,集商业可用和注释于一身。最新版本的ZINC20提供了14亿个分子和新的搜索方法。ZINC按需制作库大多来自Enamine Real数据库集合。
另外,许多制药公司都建立了专有的化合物库和空间 (图1中的“专有”),例如下面会提到礼来的PLC和辉瑞的PGVL,勃林格殷格翰的BICLAIM,GSK的XXL、默克的MASSIV、Janssen的JFS和BICLAIM等。
来自阿根廷和布鲁克海文国家实验室、芝加哥大学和Rutgers大学的研究小组从23个来源聚集了大量的小分子,利用高性能计算计算这些小分子的性质,利用计算的性质训练机器学习模型,并将得到的模型用于SARS-CoV2和癌症的筛查。他们提供了42亿个分子的结构信息,预算出来的数据大小为60 TB。
Enamine的REAL、药明康德的GalaXi和OTAVA的CHEMriya,这些库都是基于库存结构单元和已证实的反应方案,它们可分两种形式:一个巨大的化学空间 (在最近发布的REAL空间的情况下为230亿化合物) 和一个枚举的较小的化学空间子集 (例如,REAL数据库)。大型组合化学空间的构建和相似搜索将在后面讨论。
KnowledgeSpace是一个以文献为基础的数千亿虚拟产品的化学空间,具有合理的可合成性。该空间由100多个文献反应方案构成,包括环封闭、偶联、取代和多组分反应。
DNA编码库 (DELs) 在本综述中与其他文库相比并不是真正意义上的“化合物库”,但由于其规模巨大而被收录在这里。它们是由列举的分子组成的集合,分别与特定的DNA标签偶联,作为可扩增的识别条形码。由于DNA兼容反应、选择方法、下一代测序和数据分析等方面的进展,DEL技术使得构建和筛选规模空前的文库成为可能,进而帮助高效配体的发现。HitGen、药明康德和X-Chem提供DEL服务。药明康德声称其DEL含有900亿化合物。X-Chem则对外宣传有2500亿化合物。HitGen的DELs目前包含了超过1万亿种药物样化合物,这些化合物是通过精心挑选的易于处理的结构单元。
将DELs与作为本综述主要主题的虚拟库和空间区分开来很重要。除其他区别外,DEL的筛选方法不同,它们往往含有相当大的、更灵活的化合物 (例如多肽),而且它们的化学空间复盖范围相对较窄,以及更灵敏的分析读数。此外,DEL技术的全面覆盖还需要讨论噬菌体和核糖体展示以及其他技术,所有这些技术都超出了本综述的范围。
超大化合物库检索
“类比目录 (Analogue-by-catalog)”是一种基于分子相似性原理的药物发现途径,以识别易得的、与SARs可探索的相似化合物。遗憾的是,二维子结构和相似性搜索对于1亿个分子或更多的库往往无法完成,但最近出现了一些快速搜索大型库的方法。尽管其中有些内容在同行评议的文献中没有讨论过,但我们认为这里值得提及。如果本节未给出引文,读者应谨慎对待信息,因为信息已通过口头或软件论证收集。表1总结了数据库搜索技术的一些实例。
表1.超大数据库搜索技术示例

数十年来,数据库盒 (database cartridges) 一直是在标准关系数据库中添加化学特定功能的技术解决方案。最近,XChemistry在纯SQL中进行了子结构和上层结构搜索的实验,通过显式脚本路径执行逐原子匹配,使用临时的、瞬态的表。结构连接性存储在简单的一对原子和键表中,而不是任何标准格式 (例如 SD文件) 或预备盒式二进制Blob对象。各种查询格式 (例如,SMARTS、 QuerySLN、QueryMolfile或QueryCDX) 在数据库外部进行预处理,解析为共同的内部表示,然后转化为相当复杂的SQL查询语句。递归SMARTS、立体化学、R-group搜索和某些其他特性尚未实现。Xemistry已经证明,180万个复合数据集上的许多典型的纯SQL子结构查询可以在工作站PC上完成,PC上有一个标准的、未分片的、单线程的数据库 (例如MySQL或PostgreSQL) 在< 1s内完成。(分片是一种将大型数据库拆分成较小组件的水平划分方式,这种划分方式速度更快,更容易管理)。
在化学数据库中进行相似性搜索通常是将分子表示为指纹,以Tanimoto系数作为相似性度量。这是化学信息学指纹分析平台chemfp所采取的方法,该平台包括命令行工具和用于指纹生成和高性能相似度搜索的Python库。在一个应用中,指纹压缩和切分已经与chemfp数据库一起使用。Probst等人报告了一种新的指纹MHFP6,它提取了每个原子周围所有圆形子结构的SMILES,其直径可达6个键,并将最小哈希方法应用到所得到的集合中,提高了精确最近邻搜索的性能,并使局部敏感哈希 (LSH) 近似最近邻搜索算法得以应用。
MolSoft的Giga-Search可以使用SMILES和SMARTS搜索EnamineReal数据库。该方法为MolCart搜索引擎增加了指纹位统计,同时也提供了一种新的、高效的存储化学指纹的方法,以尽量减少服务器端需要扫描的数据量。Giga-Search在Enamine REAL 2020和SAVI 2020上进行了测试。据报道,在5个相当简单的子结构搜索中获得第一个1000个hits的时间在3.66到4.21秒之间变化。
OpenEye软件对其指纹图像搜索软件进行了改进,在Orion的分子服务 (MaaS) 模块中,可以将数据库存储在云中,并预测出指纹。从Enamine REAL中搜索8亿个分子的二维相似度需要3 s左右。然而,要保存多个大型数据库,需要最大的亚马逊云服务器的配置为48个物理核心上的768GB内存和96个逻辑处理器。
Schrödinger通过一个名为GPU Similarity的包含工具,在LiveDesign中提供了一个专有的、非常快速的相似搜索。由Schrödinger拥有和操作的GPU驱动的服务器托管的商业可用的化合物库包含了大约16亿个化合物。
Chemaxon的MadFastJava应用程序是一个用于超快化学相似性搜索的高端工具包。它依赖于一个优化的多线程实现和内存中的数据存储。快速的指纹生成和较短的初始化时间,伴随着大量的比较方法,允许优化相似度空间。结构以每分钟100万左右的速度输入,内存使用量约为每百万250 ~ 350 MB。MadFast在一个特定服务器上检索一亿个分子的40个相似性结构,只需大约5 s就能完成。
Google BigQuery提供了访问基于云的大型关系数据库的权限。它可以在数十秒内处理数十亿行和数十兆字节的数据,并以极低的代价将数据缩放到数百兆字节。Google BigQuery中科学数据集的可用性为利用KNIME分析平台或OntoChem的SciWalker对公共生命科学数据进行高效的探索和分析提供了新的可能。SciWalker是一种新的搜索资源,具有通过本体对知识概念进行规范化访问和分析公共和私有领域内容的能力。它利用一个分配本体概念标识符的注册系统,对大批量的科技文档进行标注和索引,这些标识符为被标注的数据提供进一步的价值。SciWalker将基于结构的化学本体与其他本体集成在一起,从而改善了科学内容的关联和分类。一旦被识别,化学实体就转化为SMILES和InChIKeys,使文档可以通过结构、子结构或结构相似度进行搜索。
Oprea和他的同事们发展了一种穷举生成支架拓扑的算法和一种有效的图的比较方法。拓扑是一个图,每个节点都有3条或4条关联边。支架拓扑的独特表征可用于识别化学子空间,并可能以更高效的方式查询大型化学数据库。
化学空间中的组合搜索
表2总结了一些组合搜索的例子。早期的基于配体的de novo设计方法包括将数据库分割成独特的结构单元,这些结构单元可以重组成新的分子。尽管由此产生的分子是有“化学感觉的 (chemically sensible)”,但合成的可及性仍然是一大挑战。为解决这一关键问题,考虑分子片段并以系统方式组合它们的方法,可以在具体的反应方案中应用。十多年前,辉瑞公司从实验室内部的笔记本中系统地解释了PGVL空间。更多的努力使其他组织取得了进一步的显著进展 (注意图1中的"专有"集合)。
表2. 超大化合物库组合搜索示例

PGVL背后的化学信息学引擎是CoLibri的早期版本,它是一个工具包,将合成知识转化为所谓的化学片段空间,以枚举格式构成大量的虚拟化合物。化学碎片空间由分子碎片和相应的连接规则组成。CoLibri反应合成器将反应定义作为输入,为每个反应定义生成一个单独的碎片空间。CoLibri碎片空间合并接收反应合成器的输出,并创建一个可搜索的碎片空间。最近,一个创建化学空间的开源工具包已经发布。
第一个用于碎片空间搜索的方法是FTrees-FS。它基于特征树 (Feature Trees),一种基于药效团的相似度搜索方法,使用了一种简化的图表示。FTrees-FS不是通过数十亿个化合物来枚举空间和筛选,而是直接从适合查询的空间构造分子。搜索的结果是一个类似于查询的组件列表,但另外,这些hit的核心片段的名称也参考了如何合成这些分子的协议。因此,化合物是通过已知的组合合成协议可以访问的。这也意味着它不仅容易合成单个分子,而且容易合成一系列类似的化合物。
图1所示的许多空间已经用CoLibri开发出来,并且可以用FTrees-FS方法进行搜索。InfiniSee是一种利用图形用户界面导航化学空间的相关解决方案。FTrees-FS和infiniSee因此都得到了广泛的应用。这种用于从浩瀚的组合空间中挖掘相关分子的过程被称为‘SAR-bySpace’。
在相关工作中,礼来公司的科学家们引入了AsmSearch在最近的PLC中进行高效的相似性搜索。PLC的目的是首先定义可以利用礼来内部资源合成的小的类药化合物的化学空间,然后为了药物发现的目的方便在这个空间中获取化合物。PLC化学信息学引擎将礼来自动化系统上常见的一组强健反应与可用的构建块结合起来,定义了易于合成的化合物空间。基于不对称 (Tversky) 相似度的方法AsmSearch最初计算PLC中反应物和查询分子基于原子路径的指纹。非对称搜索用于选择与查询分子相似度高的反应物,因为这些反应物可能生成与查询分子相似的产物分子。在最后一步中,所有选定的反应物都经过PLC反应处理,生成可合成的化学结构。由此得到的结构集,一般在几千个范围内,可以经过正常的相似搜索过程,选择足够相似的PLC化合物。
最近发展起来的一种搜索算法SpaceLight在大规模组合片段空间中进行拓扑指纹相似性搜索。与传统的指纹方法相比,SpaceLight在保持与ECFP等分子指纹描述分子相似性的高度相关性的同时,能够利用这些空间的组合特征进行效率提升。由此生成的软件能够在标准桌面计算机上以200多亿化合物秒的速度搜索REAL等空间。与FTrees-FS一样,SpaceLight尺度上涉及到的片段数而不是产物数,因此甚至可以搜索到超过1020个分子的化学空间。
3D检索方法
美国阿斯利康和汉堡大学的计算分子设计小组探索利用遗传算子直接从化学空间操纵化合物。有了合适的适应度函数,3D中的直接优化便应运而生。在最近与法国施维雅公司的合作中,汉堡团队分析了基于形状的定向描述符射线体积矩阵在片段生长中的性能,为快速探索片段库的筛选打开了大门。
表3总结了一些通过分子对接或形状进行三维搜索的例子。
表3.通过分子对接和形状进行3D检索的案例

MolSoft的快速等距化合物物发现引擎RIDE是一种基于原子属性场的快速三维分子相似性搜索方法。它在化合物构象异构体的数据库中搜索与查询等排的分子。它是一种多构象、最优的3D叠加,其中查询分子和数据库中的分子都以多个低能构象表示,以反映两个分子的柔性本质。SAVI团队证实,RIDE可利用GPU筛选高达50万个化学构象,SAVI整体可在数小时内进行筛选。然而,在实际操作中,研究小组往往希望筛选的不是单个分子,而是基于多个活性分子的模板,这样的搜索相应地较慢。
在Orion云计算平台上,一个利用3D相似度 (FastROCS) 和自动化评估工作流程的高通量虚拟筛选项目允许产生和搜索数十亿个虚拟分子,从而可以访问一个庞大的、列举的3D虚拟化学库。FastROCS的供应商OpenEye告知我们伊马替尼的形状和特征匹配案例,例如,在AWS GPU的实例上 (2个GPU、32个虚拟CPU和16GB GPU内存),3D版本中5000万个分子的形状和特征匹配大约需要1分钟。使用几百个GPU自动地在AWS服务器上搜索25亿个虚拟分子通常需要不到30分钟的时间,3D 构象和形状是预先计算和存储的。
Yang等人与Schrödinger合作,报告了一种基于主动学习的机器学习增强分子对接协议,以显著提高传统对接的吞吐量。Top化合物被自动重排,几乎所有通过穷举对接找到的高评分支架都被捕获。Schrödinger与Enamine、MilliporeSigma、MolPort和Mcule合作,提供了可购买化合物的准备数据库,包括:碎片、类先导、半类似药物的化合物和类似药物的化合物,以便进行虚拟筛选。
VirtualFlow是一种开源的工作流,可以利用多种对接程序高效地制备和筛选超大型化合物库。它已被用于制备可自由使用的、即插即用的配体库,拥有超过14亿个市售的分子,并鉴定出一系列结构多样的与KEAP1低微摩尔亲和力结合的分子。
Atomwise的AtomNet是一个深度卷积神经网络,用于预测蛋白质-配体复合物的分子结合亲和力。它表示一个蛋白质-配体对,作为包含碳、氧、氮等通道的三维体积像素集合。该模型能够自主学习控制分子结合的特征。最近,Atomwise引入了一种利用更高效的模型 (渔船模型) 来预测AtomNet得分。先通过该模型筛选上亿化合物,然后在AtomNet上最具潜力的的化合物进行评价,这些分数被用来迭代地改进渔船模型。其基本思想是建立一个基于配体的目标特异性模型来预测得分,并且在AtomNet模型上只对这个"代理"模型的最高预测得分。AtomNet已用于Enamine库中10~160亿个化合物的38个靶点和57个筛选项目,并在实验最终中发现了hit化合物。
BioSolveIT公司进行了化学空间对接,进行基于三维结构的设计。它利用形式化的结构之间基于反应的连接,反映真实的化学。该算法遵循传统的锚定-增长策略,从最初的基于结构的de novo设计方法 (如LUDI) 发展到后来在组合库环境下的探索。一个BioSolveIT应用程序涉及对接所有REAL 空间结构单元,选择最佳100个结构,枚举1731819个化合物,对接,评分2292975个姿势,检查最佳5万个姿势,最终得到了13个候选化合物。最近,V-SYNTHES被报道可以按照同样的策略对超过110亿个化合物的REAL空间进行基于层次结构的筛选。该方法首先确定最佳骨架-合成子组合为适合进一步生长的种子,然后迭代阐述这些种子,以选择对接分数最佳的完整分子。这种方法能够在巨大的化学空间中快速检测出得分最好的化合物,同时只对实际化合物的一小部分进行对接。
从头设计到分子生成
传统的基于配体的de novo设计方法包括从大数据收集的分子碎片化和从碎片生成结构。结构生成器中的组合爆炸增长,可以通过启发式、直觉和机器学习模型来防止。本综述早些时候提到的另一个挑战是确保新分子既具有药物性质,又具有可合成性。基于反应的de novo设计系统使用易得的结构和健壮的反应,如前所述的“按需”化合物库。这使得软件能够为每个设计的化合物提出合成路线。化学语言模型 (SMILES-to-SMILES) 使de novo药物设计不需要显式的分子构建规则,他们通过学习化学的规律来生成分子,其他更近的方法也采用深度学习。
化合物的逆合成分析在计算化学中有着悠久的传统,近年来由于新的机器学习方法的兴起而引起了人们的广泛关注。基于反应的设计系统DOGS与化学高级模板搜索 (CATS) 度量相结合,研究了DOGS对已知药物的药效相似度。
Schrödinger的PathFinder采用逆向合成分析,然后进行组合合成,在综合可及的化学空间中生成新化合物。利用路径查找器驱动的复合生成、基于云的自由能扰动模拟和主动学习等方法快速优化R基团并生成新的核。与大规模枚举相比,该方法在目标导向的生成机器学习的基础上得到了增强,使得有效思想更加丰富,同时保持在预定的药物相像性属性空间范围内。
DNA编码库的分析
实现文库大小和分子性质的合理的平衡对DEL技术是一个挑战,假阳性和假阴性也是如此,分析DEL检测结果也具有挑战性。解决这些问题将有利于DEL活性物质作为药物化学的出发点。
最近,辉瑞公司报告了一种通过选择特定设计中使用的单体子集来优化库属性的算法。Martin等人系统地研究了如何增加DELs的多样性和改善可覆盖的分子性质空间。他们开发并应用了eDESIGNER这种算法,它综合地生成所有可能的库设计,枚举和概要地从每个库中抽取样本,并对它们进行评价,以选出待合成的库。
Kontijevskis发展了一种基于Bemis-Murcko支架的简化分子框架 (RCMF) 方法,作为一种抽象且非常通用的化学结构表征方式。分子由一个文本字符串表示,它为环系统、链接器和环系统之间的角度提供唯一标识符。由于表示的通用性,大量的分子会映射到同一个RCMF上,使得非常大的库 (超过1012) 可以映射成热图进行比较和多样性分析。利用RMCF生成的图谱,将多个DNA编码文库的化学空间覆盖度与ChEMBL和PubChem中药物样化合物覆盖的化学空间覆盖度进行比较。
Zabolotna等利用生成地形图 (GTM) 分析了DELs的超大化学空间。它们利用商业上可利用的结构单元生成了大约2500个DEL,产生了25亿个DEL化合物,与ChEMBL的生物相关化合物相比,这些化合物可以选择几个覆盖ChEMBL化学空间的最优DEL。
化学空间可视化
化学科学正在产生大量前所未有的包含化学结构和相关性质的大型高维数据集。需要算法对这些数据进行可视化,同时保留全局特征和局部特征,并具有足够的细节层次,以便于人类的检验和解释。表4总结了一些可视化技术的实例。
表4. 可视化技术案例

分子量子数 (MQNs) 是原子、键、极性基团和拓扑特征的计数。42种MQN描述符描述了分子的大小、刚性和极性。MQN映射是由MQN定义的42维属性空间的投影。雷蒙德的团队利用MQN maps对GDB进行可视化和搜索。FUn框架由客户端和服务器模块组成,有助于创建基于Web的、交互式的大数据集三维可视化。
Pharosis是一个起源于照明可药基因组 (IDG) 计划的网络资源,它建立在目标中央资源数据库 (TCRD) 之上,是新墨西哥大学管理的人类蛋白质组综合数据目录。利用Pharos科学家可以通过整个可药基因组进行排序和筛选,并生成诸如圆圈图表和放射状图等进行可视化。
自动化实验室
我们现在将讨论自动化合成方法,在本节中忽略DEL。
在选择或设计了若干待合成的化合物后,下一步是合成或从供应商处订购。制药企业和供应商都设计了应用程序来实现这一过程的自动化。以医药企业为例,此类应用首先会检查公司的内部库存情况。如果需要合成大量的化合物,拥有高通量化学设施的公司可能比那些需要与CRO或供应商订货的公司具有优势。
理想的自动化合成平台将能够规划自己的合成路线,并在有利于生产目标规模化的条件下执行。化学发展过程中的各个要素 (设计、路线开发、实验配置和执行) 在以往的研究中已经被精简,但是没有人提出一条融合计算机辅助合成规划、专家提炼化学方法生成和机器人执行化学合成的道路。Jensen的团队报告了一个关于完全自主合成的概念的证明,并最近回顾了自主合成的进展和展望。
在制药行业,礼来公司是最早进行自动化合成的公司之一。在DMTA循环中,从开始到结束都需要高度集成的功能和过程。考虑到这一点,礼来创建了一个集成的、全球可访问的自动化化学合成实验室 (automated chemical synthesis laboratory, ASL) 和一个自动化净化实验室。在ASL成功的基础上,L2S2 (Lilly Life Science Studio) 设计实现了一个计算驱动的DMTA循环,物理上集成了药物发现过程的多个领域。这个自动化的、基于云的平台由16个自主的、互联互通的自动化工作站组成,用于化合物和试剂管理、合成、纯化和分析、生物、生物物物理测试等功能。礼来于2019年在L2S2上验证了5个流行的化合物,此后又根据项目需求增加了几个化合物的测试。同时,还开发了生物测试能力,在L2S2上提供了几种可在同一平台上进行合成和测试的分析方法。L2S2下一代药物发现平台背后的范式是“从想法到数据”。
药物发现的闭环的主要缺失环节是从理论合成路线转向可操作的合成执行的配方:构建结构ID和源、反应条件、详细的合成指令 (即程序或配方) 和完整的自动化系统执行顺序 (进行专家审查,而不是执行)。需要考虑的因素有程序 (例如来自电子实验室笔记本)、反应条件和路线实例化 (翻译为机器可读的合成工作流)。
礼来的“CChemPrint”软件是一种化学上下文感知的、基于数百万个可用反应建立起来的数据驱动方法,用来推荐与先例衍生模板相匹配的合成路线。该系统充分利用了公司合成历史数据库中详细、稳健的反应数据,并不断更新。CChemPrint使用一个注释的反应存储库,其中使用NextMove软件的的NameRxn开发了反应数据库和本体。使用本体实现大约200万个反应被归类为700多个反应类型。其中包括220多名研究人员使用自动化合成协议在ASL系统中执行的6万多个化学反应。该方法再加上现代自动化合成平台,如L2S2,以及可利用的结构单元集合,使得药物发现研究者能够为易于解释和实现的目标化合物识别路线。为了方便路线实例化,CChemPrint借鉴反应模板的过程细节来推荐起始条件和合成协议。目标是为礼来的化学家提供经过专家评审而不是专家定义的反应路线。
我们正在接近自动化研发的可能性,但丢失的中间环节仍然是机器语言中的实例的路径化。目前,许多地方已经建立了专门的自动化合成仪器,用于多肽和寡核苷酸的合成、流动化学、糖化学、交叉偶联和放射合成。礼来公司合成实验室更广泛的应用在于它涵盖了整个实验室,但礼来的系统并不具有通用性,所使用的代码仅限于所建立的精确硬件和软件。相比之下,Cronin团队设计的反应软件、计算化学和合成化学语言在体系结构、语言和硬件要求方面具有普遍适用性。
Cronin团队开发了一个化学状态机(以前叫做chemputer):一种基于化学编程语言 XDL 合成有机化合物的自主编译器和机器人实验室平台,是一个标准系统。据报道,它可以与任何机器人系统兼容,并将与开放反应数据库、实验室自动化标准化、优化和机器学习程序以及MDL反应文件标准相连接。在一个单独的倡议中,结构化产品标签 (SPL),一个强大但相当轻的公共XML文档标准,已被扩展到化学反应 ("反应SPL")。这个扩展是为了支持化学制造过程。
机遇和挑战
Walters指出了一些涉及基于配体和基于结构的虚拟筛选的实际问题。一个是假阳性问题,在100万个分子的库上建立一个假阳性率为1%的虚拟筛选,将产生1万个预测为活性的非活性分子。另外一个问题是处理所需的时间和资源。Walters期待基于利用机器学习和化学空间的连续表示的模型的更新换代。
我们认为,由于当今庞大的按需制作的复合库所包含的化合物数量庞大,现在虚拟筛选的经典范式失效,组合搜索迟早会取代虚拟筛选,它是唯一能够处理大量按需制造的化合物集合的计算方法。超大型化合物集合的可获得性不应仅仅认为是计算上的挑战,它们是加速DMTA周期的巨大机遇。毕竟相似度以及最大公共子结构搜索的算法已经被开发出来。对于形状搜索,可以认为问题的解决达到了完整的构象采样水平;对于对接,可以认为问题的解决达到了完整的构象采样和可靠的评分水平 (表5)。但是,对于超过1010个化合物的三维搜索,除了交互式和启发式的选择,仍然是一个巨大的挑战。
表5. 先导化合物识别方法和化学空间
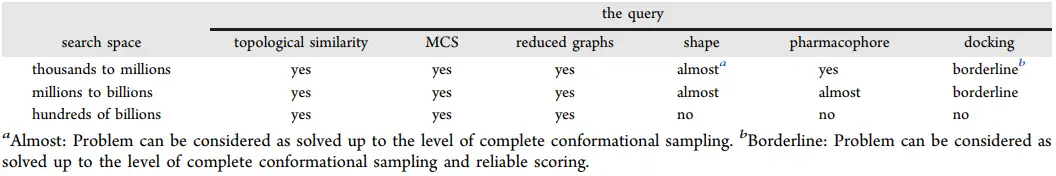
大型化合物库一旦变得更大 (例如, GDB-20),在搜索和可视化方面将面临巨大挑战。最后一次释放的REAL空间将总分子数推至约230亿,但这仍仅在1010数量级。如图1所示,实际上已经创建了超过1025个分子的空间。尽管新的算法已经开发出来 (如上所述),但是我们的眼睛和大脑将如何从可视化中提取出有意义的数据概念呢?
过去的25年被高通量、超高通量以及虚拟筛选主导。我们现在也许能够把这些技术带来的局限性抛在身后,基于片段的药物发现和具有各自软件的按需制作的化合物库使我们可以搜索生物活性物质而不是为它们筛选。越来越好的搜索工具正在开发中,但在化学空间的三维计算运用上,仍然存在诸多挑战。
参考资料
Warr WA, Nicklaus MC, Nicolaou CA, Rarey M. Exploration of Ultralarge Compound Collections for Drug Discovery. J Chem Inf Model. 2022 May 9;62(9):2021-2034. doi: 10.1021/acs.jcim.2c00224.