前言
血脑屏障(BBB)是中枢神经系统药物进入大脑发挥药效的主要障碍。如何提高小分子药物的血脑屏障透过能力一直是药物研究的主要内容之一。2021年9月中国药科大学的孙昊鹏教授等在JMC上发表综述,总结了近五年来通过结构修饰以提高小分子药物血脑屏障穿透能力的策略,希望在未来的中枢神经系统(CNS)小分子药物开发中发挥更大的作用。
介绍
中枢神经系统疾病(如阿尔兹海默症,帕金森病,脑卒中和脑癌等)是全球引起死亡的第二大疾病,严重危害人类健康。中枢神经系统疾病的治疗药物需要进入大脑发挥作用,但血脑屏障(Blood–Brain Barrier, BBB)的存在限制了药物的通过,成为中枢神经系统药物治疗的瓶颈。
血脑屏障是介于血液和脑组织之间的天然屏障,由脑微血管内皮细胞(Brain Microvascular Endothelial Cells, BMECs)、周细胞和星形胶质细胞终足组成。内皮细胞之间形成紧密连接,几乎只有小的亲脂分子能以被动扩散的方式进入。除了被动扩散之外,大脑和外界物质交换的常见机制还有主动转运、外排转运和载体介导的转胞吞作用(Carrier-Mediated Transcytosis, CMT)等。

血脑屏障对维持脑内微环境稳态至关重要,阻止亲水物质、带电分子、蛋白质和多肽进入大脑,在人体内起到保护作用,但同时也有效阻挡了超过98%的小分子药物以及几乎100%的大分子药物(如重组蛋白和单克隆抗体),使它们的脑内暴露量不足,从而限制了药效。
评估血脑屏障渗透的参数和工具
为了更好指导CNS药物设计,科学家开发了许多方法用来评价小分子的BBB渗透能力。
基于理化性质的计算机模拟
Lipinski’s五原则是基于理化性质评价化合物成药性的重要理论,同样适用于CNS药物的设计过程。基于大量实验经验,科学家提出来各种参数的参考范围。
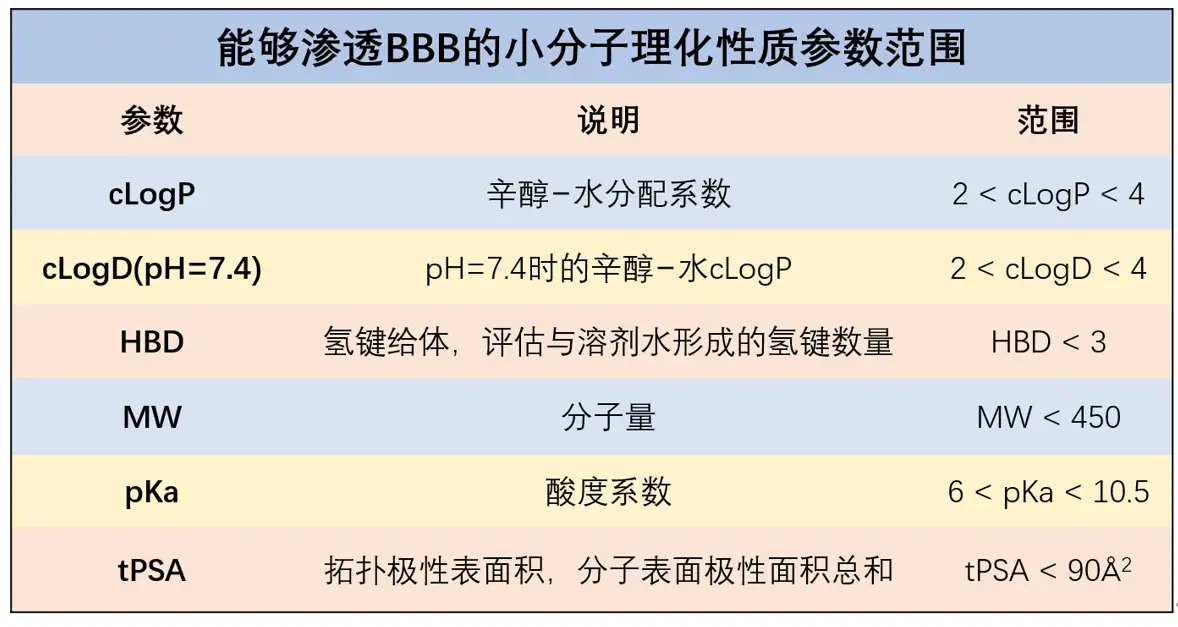
2010年辉瑞科学家开发了结合以上六个理化参数的多参数优化算法(MPO)以通过计算机模拟方法预测化合物的BBB渗透性。CNS MPO能够平衡多种可变参数,同时扩展CNS药物的设计空间且考虑了ADME(吸收、分布、代谢、排泄)对BBB渗透性的影响,此外科学家也开发出了许多其他的评价算法。所有算法的评估结果只能作为参考,具体的BBB渗透性还需要进一步的实验验证。
体外BBB渗透性预测方法
与计算机模拟相比,体外预测方法是相对准确的,而相比于体内实验,体外预测方法则更加方便。因此,体外预测方法常用于高通量筛选中以预测分子的BBB渗透性。
平行人造膜通透性分析(PAMPA)是一个研究被动吸收的渗透模型,常用于药物通过各种生物膜系统的评价,被测化合物从供槽通过单层生物膜扩散进入接收槽。人工膜通常来自包含特异性磷脂的猪脑以模拟BBB,通常以市售药物的值作为参考,以验证线性相关。以有效渗透率(Pe)表示化合物的渗透性。该法使用的人工膜并非双层膜结构且缺少转运体以至于不能反映外排和CMT情况,导致测量结果与实际情况参数偏差。
基于细胞的单层转运分析使用来自细胞的极性单层膜构建体外BBB模型,包括非脑源性的MDCK细胞、Caco-2细胞、永生化脑内皮细胞系RBE4细胞等。MDCK-MDR1和MDCK-BCRP是两个被广泛用于药物发现中评价转运体介导的外排情况的细胞系。P-gp(MDR1)和BCRP是大量表达于BBB的两个药物外排转运体,外排率(ER)是影响药物BBB渗透性的重要参数。将细胞种于96孔跨孔板(96-well transwell plates)中、在一定的高密度下形成极性单层膜,可以测定化合物从顶层转运到基底外(A-B)和从基底外转运到顶层(B-A)的转运情况。从顶层到基底外的样品被收集并且通过LC-MS分析,计算表观渗透系数(Papp)和ER。大多数的CNS药物具有很高的被动渗透性,Papp>15x10-6cm/s,当ER<2.5时表明化合物不是P-gp或者BCRP的外排底物。
体内实验测定脑内药物比率
体内评价方法具有更好的准确性和可靠性。通常使用的方法包括测定Cbrain/Cplasma参数、微透析、脑暴露评价(BEA)等方法。
Cbrain/Cplasma是广泛用于评价脑分布的参数,其值低于0.1则表明化合物不能自由透过BBB。未结合药物浓度(Cu)被认为是大脑药物发挥活性的主要部分,未结合分数(fu)是组织中可利用的游离药物比例。血浆中未结合药物浓度(Cu,p)=血浆中总药物浓度(Cp) x fu,p;脑中未结合药物浓度(Cu,b)=脑中总药物浓度(Cb) x fu,b。相应的脑-血浆未结合药物分配比(Kp,uu=AUCu,b/AUCu,p)是一个新的评价参数。Kp,uu为0.3作为一个区分脑渗透或非脑渗透临界值。微透析是一个方便测量Cu,b的方法,BEA则是以Kp,uu作为参数。
增加血脑屏障通透性或降低外排率的策略
在过去的五年中,提高CNS通透性最有效的策略依然是通过结构修饰来调整被动扩散。理化性质是决定血脑屏障穿透的关键因素。近年来的结构修饰大多基于多个参数的优化,这样在保持原有的活性的同时也能考虑到血脑屏障穿透性和成药性。
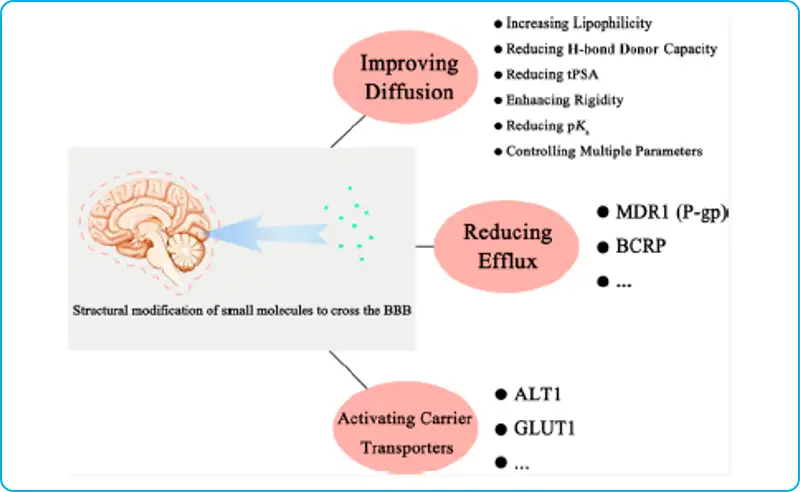
大多数情况下,血脑屏障的穿透取决于化合物的亲脂性,但由于通过血脑屏障存在多种药物转运机制,许多药物的亲脂性和药物的大脑暴露之间通常会出现偏差。由于血脑屏障的主动外排机制,许多高亲脂性药物在脑内的浓度很低。多药耐药蛋白1(Multiple Drug Resistance 1, MDR1),也称P-糖蛋白(P-glycoprotein, P-gp)转运体和乳腺癌耐药蛋白 (BreastCancer Resistance Protein, BCRP)转运体都是中枢神经系统中重要的药物外排转运蛋白。其中研究最深入的转运蛋白是P-gp,它在血脑屏障中高度表达,这也是许多药物不能进入大脑的原因。P-gp底物的结合倾向与化合物的理化性质相关。消除氢键供体、减小分子尺寸、减少电负性原子(如用S或CH2取代O)以及进行构象限制等都可以用来优化P-gp的抑制剂。
增加亲脂性
亲脂性定义为非离子化化合物在两个不混溶相(如正辛醇和水/缓冲液)中的平衡分配系数,是化合物最重要的理化性质之一。亲脂性影响化合物的吸收、分布、代谢、排泄、毒性以及药理活性等,也是一个与血脑屏障穿透性密切相关的参数。通过改善亲脂性,可以增强药物的血脑屏障穿透。在结构优化中,常用的方法是引入氟原子。氟在特定情况下的应用可以增强亲脂性,同时通过占据氧化代谢的位点提高代谢稳定性。此外,由于氟的体积小,很容易并入结构中,一般不会破坏化合物的空间构象。
间变性淋巴瘤激酶(Anaplastic Lymphoma Kinase, ALK)是治疗具有ALK基因融合或激活突变体癌症的重要靶点。克唑替尼(crizotinib,1)是广泛使用的ALK抑制剂,由于不能穿过血脑屏障,对中枢神经系统癌症转移的治疗效果较差。研究人员通过引入氟乙基增加了克唑替尼的亲脂性,血脑屏障通透性有所提高。注射5分钟后,[18F]氟乙基克唑替尼(2)的%ID/cc为6.6,而限制在血管腔内的示踪剂的预期值仅为0.2%ID/g,2可能具有更好的治疗潜力。

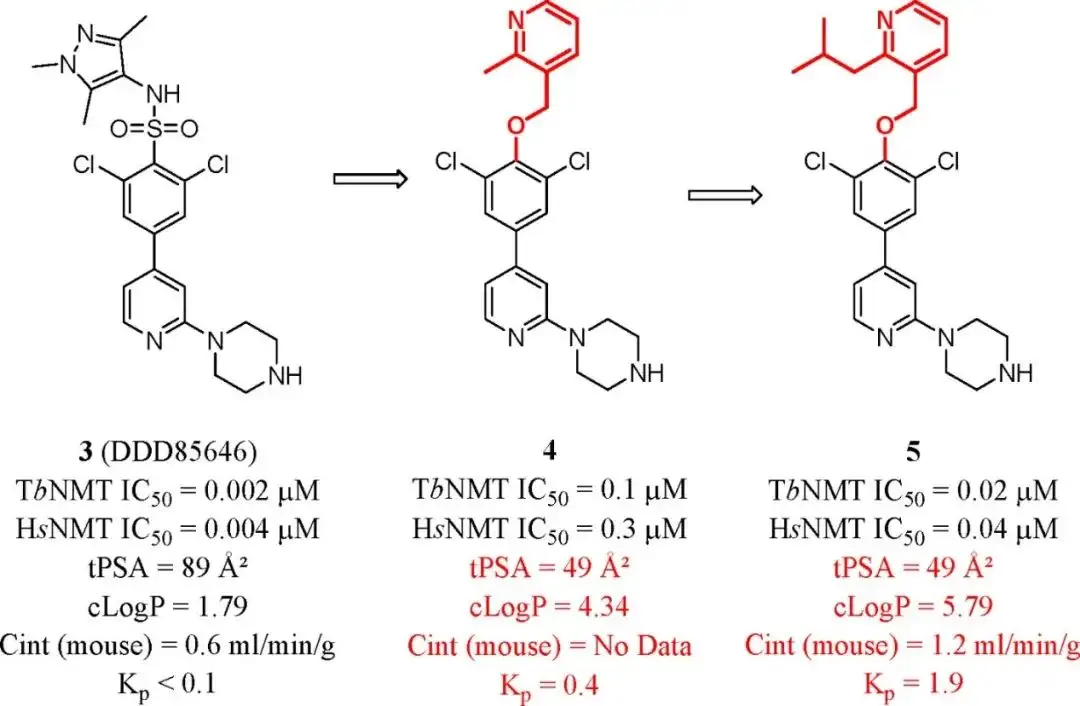
N-肉豆蔻酰基转移酶(N-myristoyltransferase,NMT) 是治疗由寄生布氏锥虫引起的人非洲锥虫病(Human African Trypanosomiasis,HAT)的理想靶点。通过高通量筛选和结构优化获得的化合物DDD85646(3)对NMT有较强的活性,但不能穿透血脑屏障(Kp<0.1)。研究人员发现3的大脑暴露量较低与较高的tPSA(89Å2)和较低的亲脂性(cLogP=1.79)有关,因此设计合成了一系列化合物3的衍生物,旨在提高血脑屏障的穿透性,其中化合物4的tPSA和cLogP得到明显改善。进一步改造以提高亲脂性,化合物5表现出明显的抑制活性、良好的微粒体稳定性和显著提高的血脑屏障穿透性。
减少氢键供体
Lipinski’s规则表明CNS药物的氢键供体(Hydrogen Bond Donor, HBD)不超过三个。减少HBD的个数已经成为提高血脑屏障穿透最常用的药物设计策略之一。研究人员用3-氯吡啶取代了6的5-氨基嘧啶,得到纤维母细胞生长因子(Fibroblast Growth Factor, FGF)受体调节剂7,改善了脑暴露量,降低了磷脂症的风险。7可能具有基础的FGF生物活性,如神经保护和细胞增殖等,可进一步开发为治疗神经退行性疾病的药物。

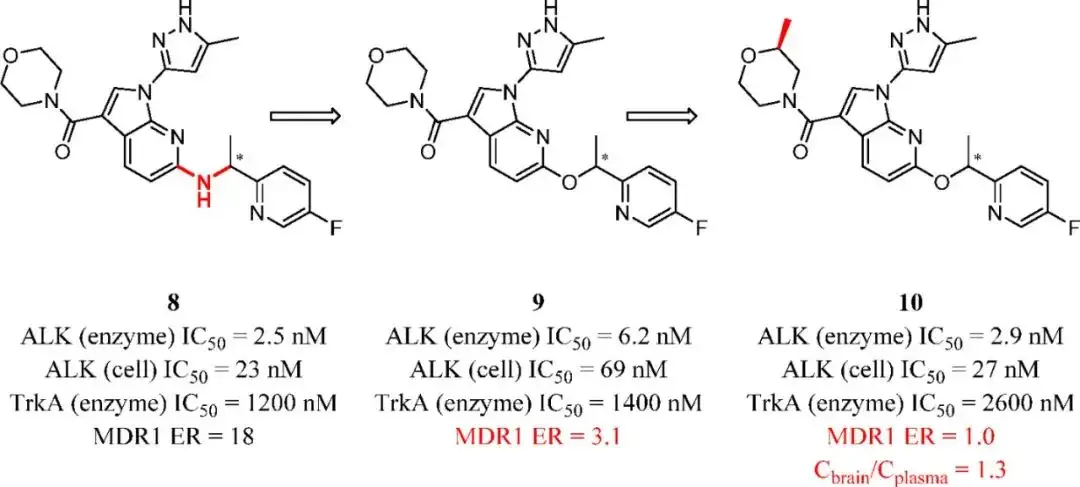
以共晶结构为指导,研究人员探索了化合物8(一种ALK抑制剂)的氢键受体和氢键供体的去除对其血脑屏障通透性的影响(这些氢键供体或受体没有与ALK活性位点形成必要的相互作用)。去除其中一个HBD得到的化合物9显著抑制MDR1外排,大脑暴露有很大改善。当甲基屏蔽了吗啡啉环上的氧原子时,MDR1的外排比进一步降低。化合物10可以作为一种体内工具来阐明ALK在认知障碍、焦虑和抑郁等中枢神经系统疾病中的作用。
当分子结构中的HBD是维持生物活性所必须时,可以考虑构建分子内氢键来提高血脑屏障通透性。化合物13具有中等的血脑屏障穿透水平,转换酚羟基的位置,使极性的羟基和叔胺基团之间形成分子内氢键,脑渗透性得到提高。化合物11和12具有抑制Aβ42纤维性颤动的生物活性和良好的膜通透性,有望成为治疗AD的多功能药物。

反之,增加HBD的数量可能有助于开发具有外周选择性的药物。KCNA5(KV1.5)基因编码的IKur在心房复极中起重要作用。化合物14有效阻滞IKur电流,是一种安全性良好的维持正常窦性心律的药物。然而,高度的脑暴露量使化合物14产生潜在的中枢神经系统副作用。研究人员发现,在引入氢键供体后,化合物15在保持IKur电流阻滞的体外活性和选择性的同时,大脑暴露率降低。

减小tPSA
tPSA定义为分子中极性原子表面的总和,与分子中极性原子数有关,是预测分子输运性质的重要参数之一,影响血脑屏障通透性、药物代谢、外排等。中枢神经系统药物tPSA值一般较低(tPSA<90 Å2),降低tPSA为改善大脑暴露提供了可行性。
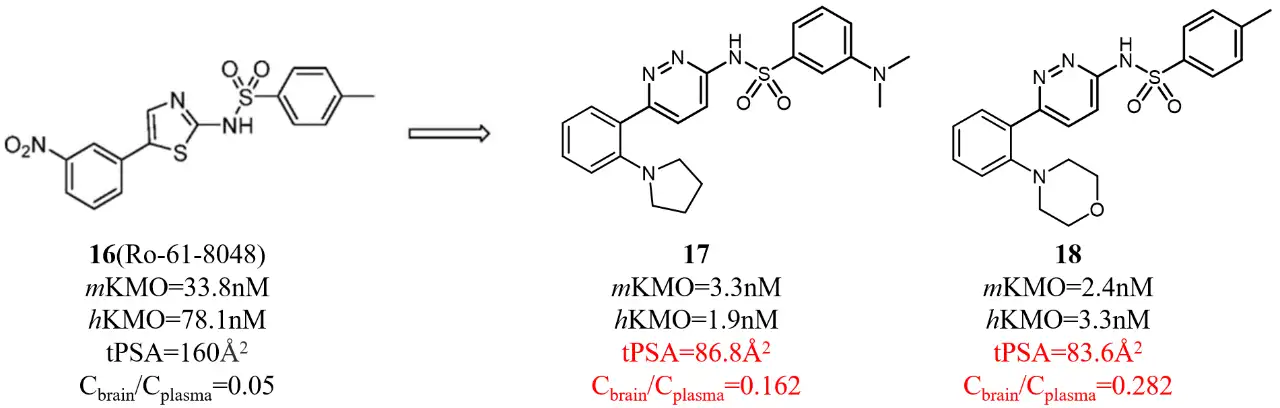
犬尿氨酸单加氧酶(Kynurenine Monoogenase, KMO)是治疗亨廷顿病(HD)的靶点。抑制KMO有望减少HD相关的毒性代谢物3-羟基犬尿素和喹啉酸。Ro-61-8048 (16)是一种脑疗效较差的KMO抑制剂。可能是由tPSA较高(160Å2)导致的。化合物库中筛选得到的化合物17具有相似的母核,显示出较好的抑制活性和适宜的tPSA值(80.3Å2)。进一步改造得到的化合物18具有纳摩尔级的活性和中度的脑暴露量。由此说明降低tPSA是提高血脑屏障穿透的有效策略。
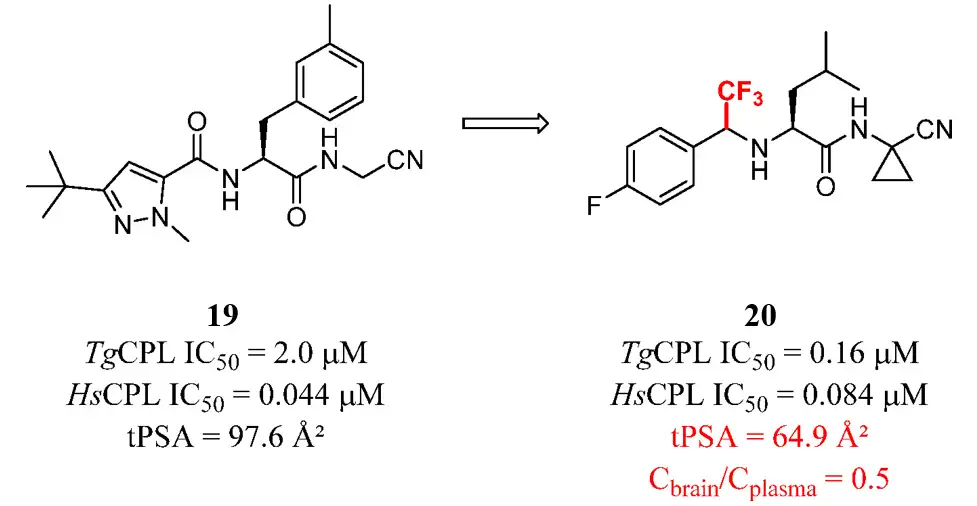
弓形虫组织蛋白酶L(Toxoplasma gondiicathepsinL, TgCPL) 是治疗寄生虫感染的潜在靶点。先导化合物19是人组织蛋白酶L(human cathepsin L, HsCPL)的抑制剂,其tPSA值(97.6Å2)较高。研究人员试图去除一些不关键的极性原子,以减少总tPSA。例如,酰胺的羰基转换为常用的电子等排体三氟甲基,化合物20显示出对弓形虫的高效活性和良好的血脑屏障穿透性。
增加分子刚性
CNS药物通常刚性较强,具有多环的体系和少量的可旋转键。Ghose等人提出,分子的柔性可以用环外直链的数量和可旋转键(Rotatable Bonds, NRB)的数量来表征,首选范围是2-4个环外直链和1-4个可旋转键。几种类药性刚性片段应用广泛,例如,苯并氮杂卓是许多中枢神经系统药物(如地西泮、加兰他明和氯卡色林)中常见的骨架。

厄洛替尼(Erlotinib,21)是第一代表皮生长因子受体(EpidermalGrowth Factor Receptor, EGFR)酪氨酸激酶抑制剂,广泛用于治疗EGFR敏感突变的晚期非小细胞肺癌患者。在近60%的胶质母细胞瘤中,表皮生长因子受体发生突变和扩增。然而,由于没有足够的脑内暴露,几乎所有的EGFR抑制剂都未能改善胶质母细胞瘤患者的预后。因此,开发高血脑屏障穿透性的新型EGFR抑制剂是十分必要的。研究人员猜想厄洛替尼的大脑穿透能力差与结构中灵活的烷基醚尾部有关,导致NRB(10)较大和tPSA(75Å2)较高。闭合烷氧基链,形成一个1,4-二氧六环,融合到喹唑啉骨架上,得到了化合物22,NRB(2)减少,tPSA(56Å2)降低。与厄洛替尼相比,化合物22的血脑屏障穿透率提高近10倍(Kp,uu=0.491)。进一步引入卤原子,JCN037 (23)的脑暴露量进一步增加(Kp,uu=1.30),在体内和体外均显示出有效的活性。

磷酸二酯酶4D(PDE4D)负责催化cAMP分解,这是记忆形成的关键步骤。抑制PDE4D已被证实有助于AD等退行性疾病的治疗。在先导化合物GEBR-7b(24)与PDE4D蛋白的对接和分子动力学模拟中,研究人员发现亚胺醚基团与PDE4D催化位点形成关键的氢键相互作用。试图引入杂环降低分子的灵活性,限制其构象,得到化合物25。在药代动力学分析中,化合物25迅速被吸收并到达大脑。与24相比,25血脑屏障穿透明显增加。

减小pKa
减小pKa也是提高血脑屏障渗透最常用的药物设计策略之一。pKa与ER有显著的相关性。
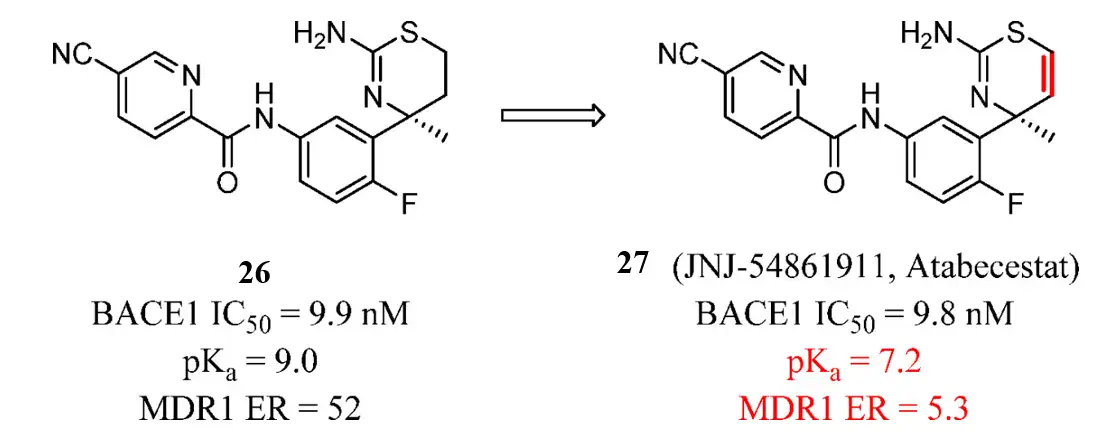
β位点淀粉样前体蛋白切割酶1(β-site amyloid precursor protein cleaving enzyme1, BACE 1)抑制剂可以阻断淀粉样β多肽(amyloidβ peptides, Aβ)合成的限速步骤。Aβ积累可能与阿尔兹海默症发病有关。抑制BACE 1有延迟甚至预防AD进展的潜力。化合物26的 P-gp外排水平高(在MDCK细胞中ER=52)。分析表明pKa<8的化合物不太可能是P-gp底物。化合物26的pKa为9.0。通过合理设计,降低pKa,JNJ-54861911(Atabecestat,27)活性保持不变,ER值降低,在大脑中的外排减少。
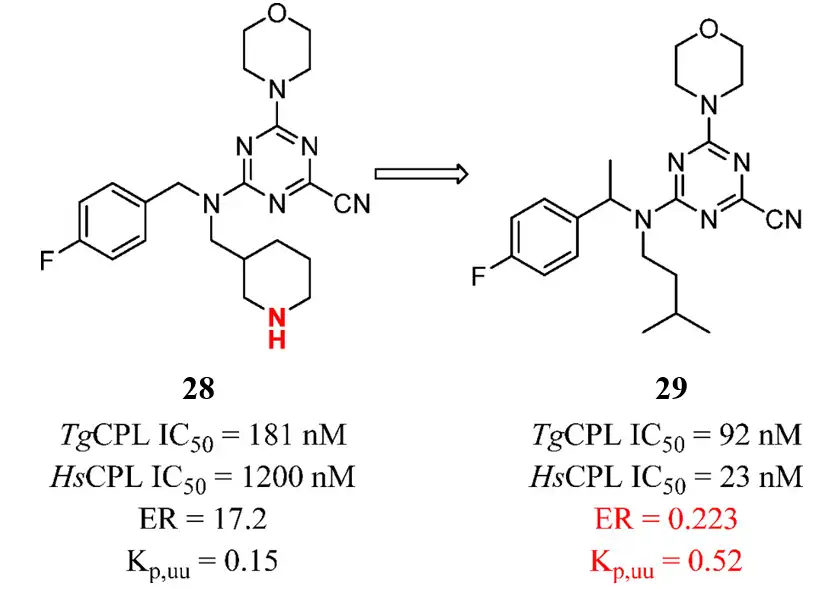
研究人员在体外评估TgCPL抑制剂28和29,以确定它们是否为P-gp的潜在底物。化合物28的高ER值解释了其脑内的低暴露量,这可能是由于结构中碱性胺的存在。

调控多参数
在多数情况下,改善血脑屏障的通透性不只是优化一个参数,而是综合使用多种优化策略。有时,单个参数的变化不能满足预期的要求,或者会导致其他重要特性的变化,如活性和药代动力学。
LY288183542(30)是一种游离脂肪酸受体1(Free FattyAcid Receptor 1, FFAR1), 也称为G蛋白偶联受体40(G protein-coupled receptor 40, GPR40)的激动剂,可能存在血脑屏障通透性低的问题。GPR40是治疗2型糖尿病的理想靶点,可以通过葡萄糖刺激胰岛素分泌和肠促胰岛素分泌两种机制来调节血糖稳态。但是GPR40也可能与镇痛、成人神经发生和神经血管退化有关。化合物30有一定的CNS暴露量(Cbrain/Cplasma=0.14)。此外,30在体外表现出高清除率(在人肝微粒体中t1/2=18min,在小鼠肝微粒体中t1/2=2min),在体内半衰期短(t1/2=1.3h)。对多个位点进行修饰,发现羰基替换增加了tPSA,降低了LogP。高刚性螺[茚-1,4'-哌啶]环的开环增强了分子的灵活性。将氮原子引入哌啶基进一步限制了分子进入大脑。化合物31和32具有较长的半衰期和良好的激动活性,血脑屏障渗透性差,基本不会产生中枢神经系统副作用。
结构简单的分子可能更容易通过血脑屏障。精简化合物的部分结构可以使其分子量更小,HBD更少,tPSA更低,但也存在活性降低的风险。化合物33为μ-阿片受体(μ-opioidreceptor, MOR)/κ-阿片受体(κ-opioidreceptor, KOR)激动剂。化合物33并非吗啡的结构类似物,可能是一种新骨架的小分子阿片受体激动剂。化合物33的一个合成中间体表现出较弱的MOR激动活性,作为先导化合物对其进行结构修饰。合成了化合物34和35,它们表现出强大的活性,EC50值为10nM。34和35的脑浓度分别约为33的5倍和8倍,这证实了简化结构有利于CNS的分布。

因此,在中枢神经系统药物的发现与设计过程中,引入额外的片段需要特别注意,比如杂原子、HBD、碱性或大分子量的基团。VU0483253(36)是一种毒蕈碱乙酰胆碱受体亚型5(M5)的负变构调节剂,研究人员旨在通过结构修饰改善其药代动力学,并保持高选择性和高CNS穿透。改造得到的化合物37消除半衰期非常短(t1/2<30分钟),活性没有增强,血脑屏障穿透也降低了。这可能是由于环丁基(甲基)氨基的引入,使分子量、tPSA和pKa同时增加。直接在氯的邻位引入一个甲基得到的VU6008667(38)在大鼠中的t1/2降低了约35倍(t1/2=2.3h),同时保持了适宜的活性和良好的中枢神经系统穿透。

利用载体介导的转胞吞作用
一些必需的营养物质如葡萄糖和氨基酸,不能通过被动扩散通过血脑屏障,需要通过载体介导的转胞吞作用(CMT)才能到达大脑。将药物修饰成转运体底物的类似结构,从而被血脑屏障上的特定转运体识别,可以显著改善大脑对药物的摄取。此外,这种策略也有效地降低了循环中游离母体药物的浓度,从而减轻脱靶效应。
利用载体介导的转胞吞作用通常是通过设计前药来实现的。前体药物需要被转运体识别并通过血脑屏障运输,然后通过酶促反应释放母体药物。一种能将药物传递到大脑的前药应具备以下特征。(1)具有足够的外周循环稳定性,使药物在进入大脑前不被破坏;(2)该结构必须被CMT蛋白识别,从而与内源营养物质竞争;(3)可以经过特异性水解,在脑内释放出母体药物;(4)安全性良好。此外,应注意前药不能影响大脑对必需营养素的摄取。
利用L型氨基酸转运体(L-Typeamino acid transporter, LAT1)进行前药的运输是最常见的设计。LAT1是一种跨膜异二聚体蛋白,由于大脑需要持续的氨基酸供应,LAT1在血脑屏障、神经元、星形胶质细胞和小胶质细胞中高度表达。临床上常用的几种氨基酸类药物和前药均通过LAT1转运,如左旋多巴(L-dopa)、加巴喷丁(gabapentin)、美法仑(melphalan)等。
研究人员设计合成了环加氧酶抑制剂酮洛芬(ketoprofen)的前药。单次腹腔注射给药25μmol/kg后,前药39和40能在脑内释放适宜浓度的母体药物,Kp,uu(分别为0.13和0.35)比酮洛芬本身(Kp,uu=0.01)高了10倍以上。

另有研究人员设计并合成了一种LAT1介导转运的六氢烟酸前体药物。化合物41提高了六氢烟酸的血脑屏障穿透能力,并表现出抗癫痫活性,有效地抑制了神经元和胶质γ-氨基丁酸(GABA)的摄取。六氢烟酸和41以15mg/kg剂量给药30分钟后,六氢烟酸组小鼠脑内未检测到六氢烟酸,而化合物41组小鼠脑内释放六氢烟酸的浓度为450nmol/g,大大增强了六氢烟酸的转运。

L-抗坏血酸(L-Ascorbic acid, AA)也可以作为脑内药物递送的载体。大脑中有两种转运蛋白可以转运AA。葡萄糖转运体1(Glucose transporter 1, GLUT1)可以运输AA的氧化形式,而Na+依赖的维生素C转运体(Na+-dependent vitamin C transporter,SVCT2)则直接将AA转运到大脑。布洛芬的前体药物42可以通过AA相关的转运体运输,具有良好的血脑屏障穿透能力,摄取效率是布洛芬的3.16倍。
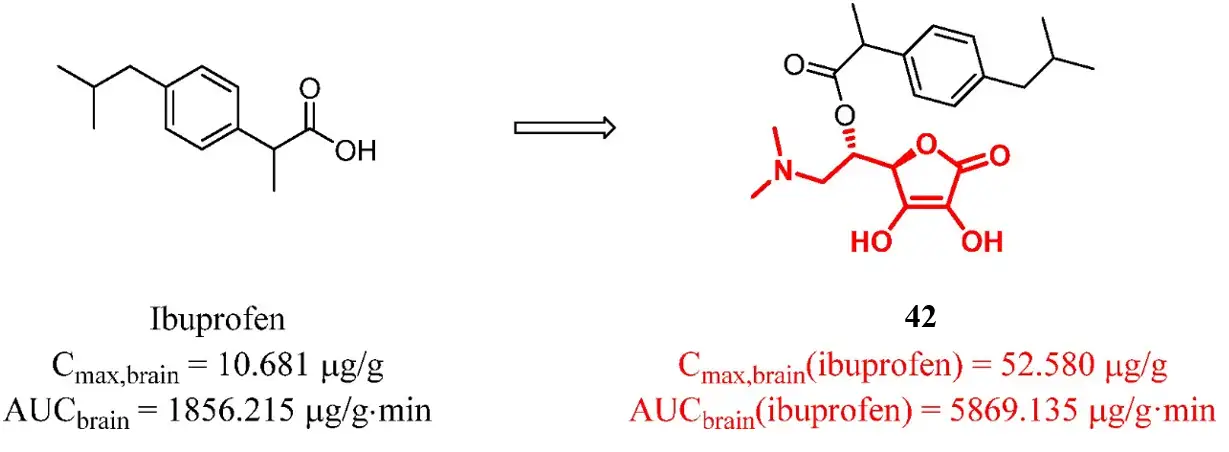
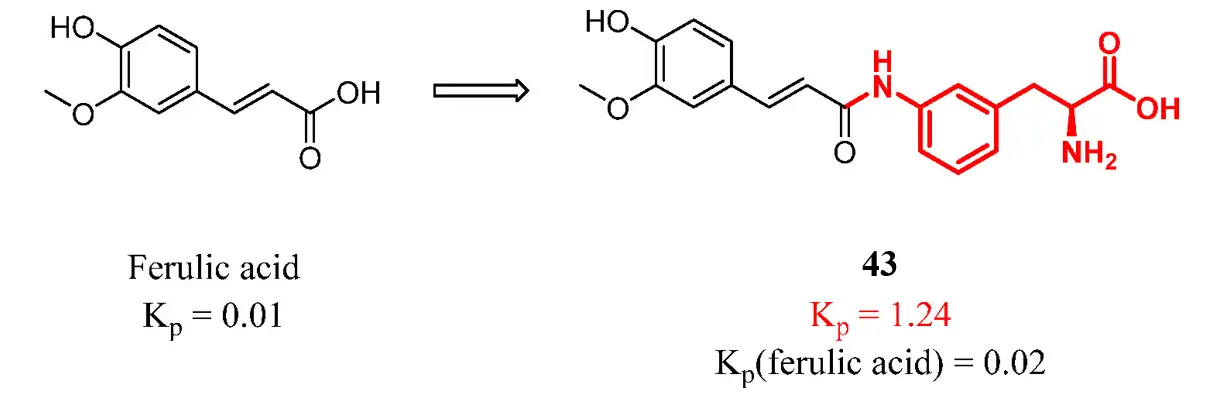
然而,这种策略并不都是成功的,前体药物不稳定,在外周循环中分解或不能在大脑中水解成母体药物都会导致大脑中的母药浓度不能得到显著增加。例如,研究人员连接阿魏酸和氨基酸残基得到前药43,43有效地与LAT1结合并穿过血脑屏障。然而,阿魏酸的脑暴露水平没有明显改善,研究人员发现,这是由于化合物43难以水解成母体药物导致的。
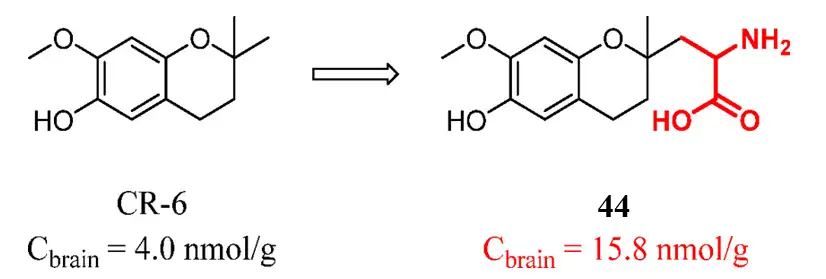
因此,另一个不同于前药的设计理念是递送载体结合到分子上并不影响靶点活性。这种策略的优点是不需要考虑前体药物的稳定性和水解性。例如,研究人员合成了一个与CR-6的内源性营养物质衍生物库,其中氨基酸衍生物44的大脑暴露量最高。44和母药CR-6通过腹腔给药,剂量为0.2mmol/kg,1h后,化合物44组Cbrain为15.8nmol/g,约为CR-6组大鼠的4倍。
中枢神经系统的特殊结构靶向
药物在全身血液循环,并通过被动扩散或主动转运进入大脑。然而,交换机制是双向的,从大脑到血液的被动扩散或主动运输同时也在进行。一些具有特殊结构的分子可以被CNS内的各种酶催化,一旦进入大脑就不能从血脑屏障运输出去。通过这种“锁定”功能,前药的运输由双向变为单向,从而提高了大脑中活性药物的浓度。
研究人员报道了基于多奈哌齐(Donepezil)的结构修饰前药。引入一个1,4-二氢吡啶环取代多奈哌齐的哌啶基团,屏蔽了氮上的正电荷。前药45由于1,4-二氢吡啶氮的良好亲脂性,能够穿过血脑屏障,而在生理pH下,它还不足以质子化。前药45在人血浆中具有良好的稳定性,进入大脑后,氧化还原酶介导的氧化还原反应将前药45转化为相应的药物46。46的N具有正电性,血脑屏障穿透度低,由此产生“锁定”效应,脑内药物浓度提高。

另有研究人员开发了一种具有“锁定”功能的脑靶向AA前体药物。布洛芬前体药物由AA载体传递,并与二硫胺传递系统结合。前药47在空气中稳定,易于保存。一旦进入中枢神经系统,它可以被二硫还原酶还原,然后环闭合生成噻唑,化合物48不能通过血脑屏障运送到脑外。48再通过水解释放活性药物,发挥治疗作用。

总结
长期以来,AD、PD和脑卒中等中枢神经系统疾病威胁着人类健康,给患者和社会带来巨大影响。然而,新的中枢神经系统药物的开发非常困难,穿透血脑屏障是开发新型中枢神经系统药物的重大挑战。
增加亲脂性、减少氢键供体、减小tPSA、增加结构刚性和减小pKa都是通过改变理化性质或降低外排来提高BBB穿透水平,此外,利用载体介导的转胞吞作用,通过一些特殊结构产生中枢神经系统“锁定”效应也有利于增强脑渗透。有时改变一个参数并不能得到预期的结果,需要结合多种策略,以达到各方面的平衡。相信在药物化学修饰的助力下,未来能有更多可供选择的中枢神经系统药物上市,为广大的患者带来福音。
参考文献:
1. Baichen Xiong, Yuanyuan Wang, et al.Strategiesfor Structural Modification of Small Molecules to Improve Blood–Brain BarrierPenetration: A Recent Perspective.J. Med. Chem.2021, 64, 18,13152–13173.
本文版权归原作者所有,文章内容不代表平台观点或立场。如有关于文章内容、版权或其他问题请与我方联系,我方将在核实情况后对相关内容做删除或保留处理!联系邮箱: yzhao@koushare.com