mTOR是一种丝氨酸/苏氨酸蛋白激酶,已被证实与多种人类疾病的发生发展密切相关。抑制mTOR信号通路可有效阻断各种生长因子的异常信号转导,从而阻断疾病的发生和发展。当前mTOR抑制剂的发展已经经历了三代,本文主要总结了mTOR抑制剂的研究进展。

mTOR简介
mTOR(Mammalian target of rapamycin)是一种高度保守的丝氨酸/苏氨酸蛋白激酶,在其C-端,mTOR与磷脂酰肌醇3-激酶(PI3K)和磷脂酰肌醇4-激酶(PI4K)的催化结构域具有高度同源性,因此属于PI3K相关激酶(PIKK)蛋白家族。
mTOR由以下6个结构域组成:HEAT结构域(Huntingtin, EF3, the A subunit of PP2A, TOR1)、FAT结构域(Focal adhesion targeting domain)、FRB结构域(FKBP12 rapamycin binding domain)、KIN结构域(Kinase domain)、NRD结构域(Negative regulatory domain)和FATC结构域(Focal adhesion targeting domain of C-terminal)。

图:mTOR的结构
mTOR可以募集其他蛋白和活性因子,组装形成两种类型的mTOR复合物,mTOR复合物1(mTORC1)和mTOR复合物2(mTORC2)。mTORC1由mTOR、RAPTOR、mLST8和两个负调控因子PRAS40、Deptor组成,mTORC2由mTOR、Protor、Rictor、Deptor、mSlN1、mLST8组成。

图:mTORC1和mTORC2的组成
mTORC1和 mTORC2在细胞中具有不同的信号传导作用。mTORC1整合有关营养丰度和环境状态的信息以调节细胞内合成代谢和分解代谢的平衡,与细胞生长有关,而 mTORC2控制细胞骨架重组,调控葡萄糖代谢,与细胞的生存和增殖有关。

图:mTORC1和mTORC2调控不同的生理活动
mTOR 主要参与四种信号通路过程,包括PI3K/AKT/mTOR、Ras/MAPK/mTORC1、Ras/MAPK/mTORC1和ULK-ATG13-FIP200通路。许多疾病的发生和发展都与mTOR信号异常有关,其中大部分表现为mTOR的过度激活或过度表达。持续过度激活的 mTOR 信号将导致细胞代谢水平的提高、持续的生长和增殖、细胞寿命延长甚至细胞永生化,这可以直接或间接诱发癌症、代谢和衰老有关的疾病。而抑制这种状态可以有效延缓或治疗mTOR过度激活引起的癌症、心血管损伤等疾病。

图:mTOR参与的信号通路
目前mTOR抑制剂主要分为三类:抗生素变构mTOR抑制剂(第一代)、ATP竞争性mTOR抑制剂(第二代)和其他新型mTOR抑制剂(第三代)。
mTOR抑制剂
第一代mTOR抑制剂
第一代mTOR抑制剂一般是指抗生素类变构mTOR抑制剂,主要是雷帕霉素(Rapamycin, 1)及其衍生物(Rapalogs)。
雷帕霉素水溶性和稳定性差,生物利用度低,对雷帕霉素进行一系列的结构修饰和改造,以期提高药代动力学。雷帕霉素的各个位点的修饰均有报道,研究最早且相对成熟的修饰位点是C-42位,这也被认为是不削弱整个化合物生物活性的最佳修饰位点。
Temsirolimus(CCI-779, 2)是惠氏公司开发的第一种雷帕霉素衍生物,通过CYP3A4转化为雷帕霉素,其水溶性和稳定性均优于雷帕霉素,但酯类口服易降解,因此Temsirolimus仅用于静脉给药。Everolimus(RAD001, 3)是诺华公司开发的一种口服雷帕霉素衍生物,也具有改善的水溶性和稳定性,对多种肿瘤的治疗有效。Ridaforolimus (AP23573, MK-8669, 4),也称为Deforolimus,由默克和 ARIAD 开发。Umirolimus (TRM-986, 5)具有高度亲脂性,在免疫抑制和抗增殖方面显示出有效活性。Zotarolimus(ABT-578, 6)在C-42位有一个四唑取代基,呈现(S)-构型,这与其他雷帕霉素衍生物相反。

图:雷帕霉素及其衍生物的结构
雷帕霉素并不直接抑制mTOR 活性,它与FK506结合蛋白12(FKBP12)结合形成FKBP12-雷帕霉素复合物,该复合物再结合mTOR的FRB结构域,其中,雷帕霉素嵌入由 FKBP12 和 mTOR 的 FRB 结构域形成的空腔中,改变mTOR的构象,从而以变构的方式抑制mTORC1活性。因此FKBP12只是雷帕霉素介导的 mTORC1 抑制的辅助因子。由于 FRB 结构域是mTOR独有的,因此雷帕霉素对mTOR具有极好的选择性并且在纳摩尔范围内有效。雷帕霉素的衍生物的作用机制与雷帕霉素类似。

图:雷帕霉素作用机制
第二代mTOR抑制剂
第二代mTOR抑制剂主要是ATP竞争性抑制剂,与mTOR激酶结构域中的ATP结合位点结合。从结构上看,可分为以下几类。
01吡唑并嘧啶类
吡唑并嘧啶是激酶抑制剂的常用母核之一。研究人员对化合物库进行筛选产生了两个4-氨基吡唑并嘧啶母核化合物S1和S2。进一步改造得到的PP242(7)和PP30(8)对mTOR有较强的选择性和活性,IC50分别为8和80nM。PP242还能在一定程度上抑制PKC、RET和JAK2的激酶活性,这可能与它的结构与ATP的腺嘌呤相似有关。PP487(9)和PP121(10)也具有一定的酪氨酸激酶抑制活性,可以抑制Abl、Hck、Src、VEGFR2和PDGFR。基于PP242改造得到的化合物MLN0128是mTORC1和mTORC2的双重抑制剂,具有浓度依赖性,抗肿瘤特性优于雷帕霉素。化合物12,13,14也属于4-氨基吡唑并嘧啶类化合物,在吡唑的两个取代基上进行改造得到。
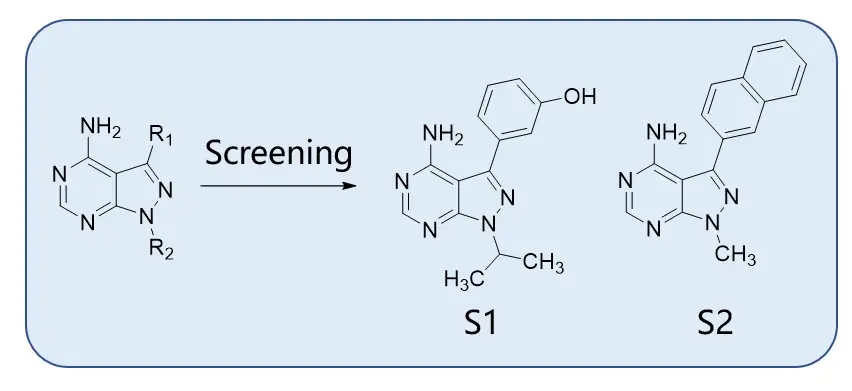

图:吡唑并嘧啶类mTOR抑制剂
惠氏公司基于高通量筛选发现的先导化合物WAY-001(15)对mTOR的IC50为220nM,对PI3Kα的效力是mTOR的6倍。WAY-001的酚羟基是一个潜在的葡萄糖醛酸化位点,用吲哚或氨基甲酸甲酯等替代苯酚,克服了这种潜在的代谢倾向,同时保留了这些基团作为氢键供体的功能。WAY-600(16)、WYE-687(17)和WYE-354(18)对 mTOR 的选择性高于PI3Kα(>100倍)和PI3Kγ(>500倍)。进一步的优化产生了WYE-132(19),对mTOR的IC50为0.19±0.07nM,能有效抑制多种癌细胞系的增殖,IC50值通常在低纳摩尔范围内。化合物20也是一种有效的,选择性的mTOR抑制剂。

图:吡唑并嘧啶类mTOR抑制剂
02咪唑并嘧啶类
基于高通量筛选发现的化合物21是一种中等有效的mTOR抑制剂,Ki值为72nM。在铰链结合区域引入取代吗啡啉,并且在嘧啶骨架上用苯基脲替换氨基嘧啶可以提高mTOR的效力及对PI3K的选择性。化合物22(mTOR Ki=9nM)对mTOR的抑制活性是21的8倍,相比于PI3Kα和PI3Kδ的选择性分别是21倍和17倍。选择性的产生是由于S-甲基吗啉对mTOR中特有的Trp2239残基的堆积作用和乙基脲与Glu2190侧链的Cβ和Cγ原子之间存在疏水相互作用。对22进行进一步优化,得到的吡唑并[4,3-d]嘧啶化合物23是该系列中最有效的化合物。此外该系列的化合物24和25均具有纳摩尔水平的mTOR抑制活性和相比于PI3Kα的高选择性。
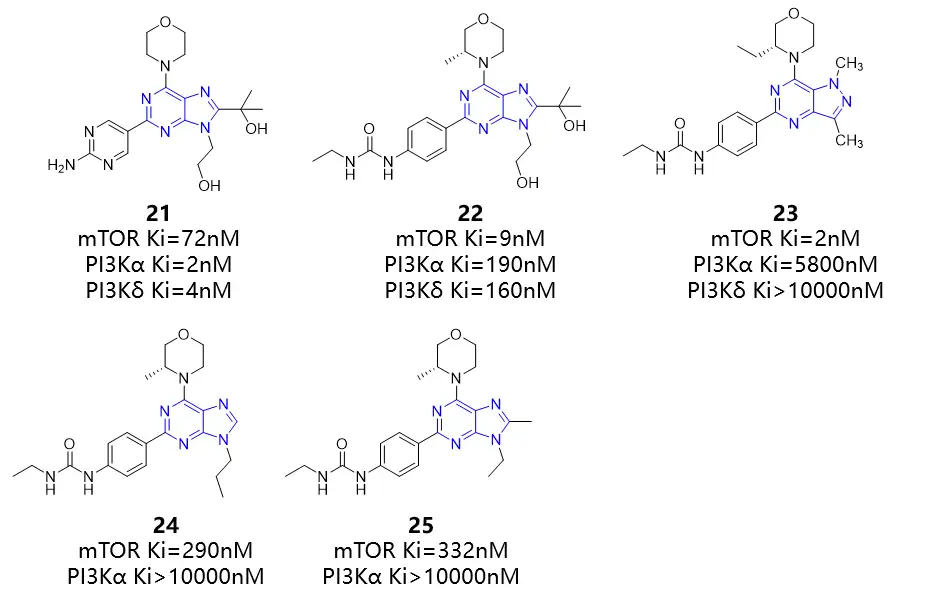
图:咪唑并嘧啶类mTOR抑制剂
03吡啶并嘧啶类
基于高通量筛选获得的外消旋化合物26具有一定mTOR抑制活性,进一步的构效关系研究发现的KU0063794(27)可以同时抑制mTORC1和mTORC2,IC50为10nM,具有高度的选择性,在10μM浓度下对其他76个蛋白激酶和7个脂质类激酶(I类PI3Kα和PI3Kβ, II类PI3K-B, III类VPS34以及SPHK-1,SPHK-2和胆碱激酶)也没有抑制活性,但该化合物相对较低的水溶性(2.4μM)和相对较高的hERG毒性(IC50=8.3μM)限制了进一步的临床发展。为改善水溶性,尝试了引入碱性基团,降低亲脂性等多种策略。AZD8055(28)是一种口服生物可利用的、有效的、选择性的 mTOR 激酶抑制剂,相比于PI3K亚型或相关PIKK 家族成员的选择性约为1000倍。AZD8055在体内外具有良好的肿瘤细胞增殖抑制活性,目前处于I期临床试验阶段,但大鼠的口服生物利用度低,仅为12%。为改善药代动力学性质,进一步的构效关系研究得到AZD2014(29),水溶性得到提高(>600μM),hERG毒性明显降低(IC50=47.5μM),大鼠的口服生物利用度提高为40%。但阿斯利康于2018年终止了处于II期研究的AZD2014。化合物30是一种新型PI3K/mTOR 双重抑制剂,对mTOR的选择性比PI3K家族高10倍以上,对乳腺癌细胞系MCF-7,MDA-MB-231和MDA-MB-468具有较高的抗增殖能力。将化合物30末端苯基脲基团上取代基的性质从氢键供体转变为氢键受体能够促进与PI3Kα蛋白的额外相互作用,增强对PI3Kα的亲和力。化合物31以剂量依赖性方式抑制MDC7 细胞增殖,IC50约为170nM。
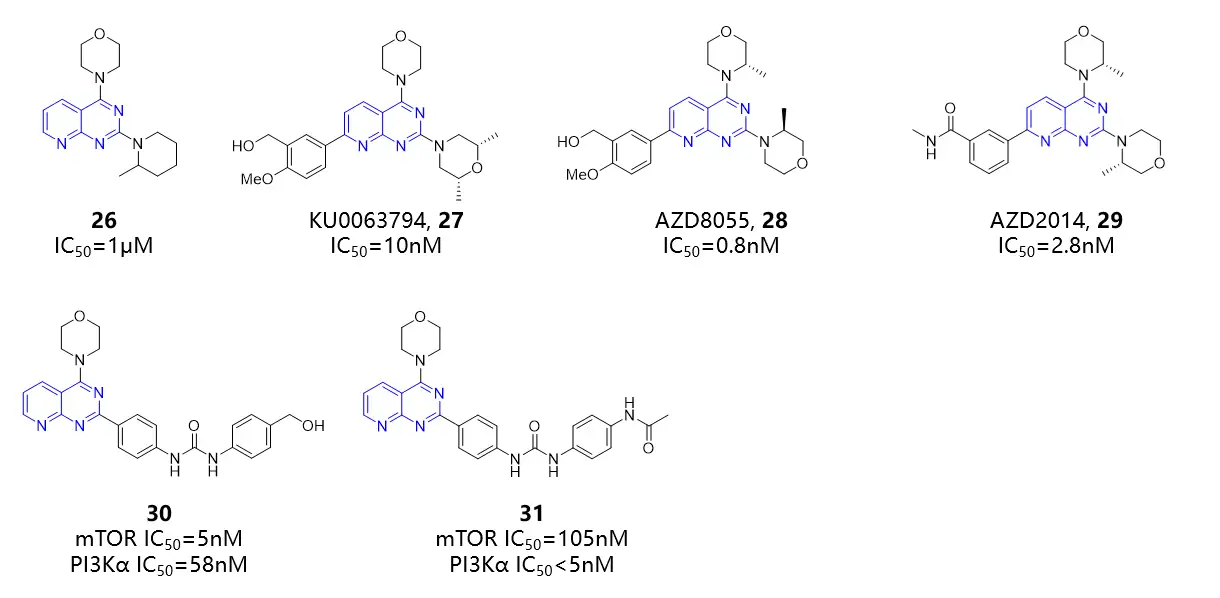
图:吡啶并嘧啶类mTOR抑制剂
04噻吩并嘧啶类
GDC-0941(32)是一种PI3K抑制剂,同时对mTOR也有一定的抑制作用,对mTOR的IC50值为580nM,对PI3Kα的IC50值为3nM。进一步研究发现用2-氨基嘧啶替代吲唑片段可提高mTOR的抑制活性,在噻吩嘧啶核上引入甲基,可降低体内清除率,用乳酸酰胺代替磺酰胺可增加化合物在中性pH下的热力学溶解度。GDC-0980(33)是一种PI3K/mTOR的双重抑制剂,对乳腺癌、胰腺癌、NSCLC和结肠癌细胞系具有优异的增殖抑制作用。在噻吩并嘧啶母核上引入桥环吗啉和苯基脲获得的化合物34是该系列中mTOR抑制活性最强的化合物,对mTOR的选择性为PI3Kα的400多倍。GNE-493(35)是基于GDC-0941的优化获得的另一个化合物,具有良好的体内药效和药代动力学特性,相比于130多种激酶具有良好的选择性。在GDC-0941噻吩环上引入甲基以破坏平面性,得到了GNE-477(36),其体内清除率有所改善。

图:噻吩并嘧啶类mTOR抑制剂
05三嗪类
PKI-587(37)是具有三嗪骨架的新型有效的PI3K/mTOR双重抑制剂,三嗪骨架中三个氮原子的存在使分子极性增大,cLogP更低。PKI-587对mTOR 的IC50为1.6nM,对PI3Kα的IC50值为0.4nM,对MDA-361和PC3-MM2细胞具有明显的生长抑制作用,在体内模型中也表现出良好的药效。研究人员将2-氧代三嗪结构引入该类抑制剂中,得到的化合物38对MDA-MB-361和PC3肿瘤细胞株明显增殖抑制作用,IC50分别为3和9nM,在大鼠、裸鼠、小鼠和人微粒体中具有高溶解性和良好的稳定性。PKI-179(39)是基于PKI-587改造而来的另一个化合物,为了增加cLogp值,将PKI-587中的一个吗啉环以桥环吗啉替代,同时保留另一个吗啉环与铰链区的Val851形成关键的氢键相互作用,此外,通过去除酰胺部分,使化合物的分子量降低到了500以下,PKI-179对mTOR的IC50为0.42nM,对PI3Kα的IC50为8nM,对MDA-361和PC3mm2细胞株有显著抑制活性。该化合物在裸鼠和大鼠体内非常稳定,但在人体内稳定性适中(t1/2=14min),代谢产物鉴定表明,桥环吗啉基团是主要的代谢位点,代谢产物40也具有一定mTOR抑制活性,IC50为0.8nM,对PI3Kα的IC50为4nM。

图:三嗪类mTOR抑制剂
ZSTK474(41)是一个具有三嗪母核的泛PI3K抑制剂,为了提高化合物的溶解度、生物利用度和血脑屏障透过率,并且适当引入对mTOR的抑制活性,改造得到了PQR309(42),对mTOR的IC50为89nM,对PI3Kα的IC50为33nM,具有快速吸收的特性,口服生物利用度良好。基于PQR309改造得到的PQR620(43)是一种新型有效的选择性mTORC1/2抑制剂,对mTOR的亲和力和选择性增加。化合物PQR620在66种癌细胞系的平均生长抑制IC50为0.92μM,口服给药后表现出良好的暴露性和优异的脑渗透性。此外,还有实验表明PQR620能缓解Tsc1GFAP敲除小鼠的癫痫发作。同样改造于PQR309的PQR514(44)细胞活性更好,OVCAR-3异种移植模型中,在浓度低于PQR309约8倍的条件下就能显示出显著的肿瘤抑制效果。

图:三嗪类mTOR抑制剂
另有研究人员通过化合物库筛选得到了化合物45,其mTOR的抑制IC50为270nM,且对其他PIKK家族有选择性(PI3Kα和ATM IC50>10μM)。考虑到腙结构具有一定的化学不稳定性,代谢也存在一定问题,因此引入环状类似物咪唑替代腙片段,得到的化合物46对 mTOR的效力显著提高,对mTOR的IC50为21nM,对PI3K的IC50大于10μM。同样由高通量筛选发现了另一类三嗪苯并咪唑母核的mTOR抑制剂化合物47对mTOR的抑制IC50为97nM,但对PI3K激酶也有活性,选择性不强。基于构效关系优化得到的化合物48活性和选择性较优,mTOR的抑制IC50为27 nM,比PI3Kα高30倍,其三嗪环上的取代基发生了变化,同时替换了可能存在药代动力学问题的苯酚基团,但48的溶解度较差,导致口服暴露低。后续研究发现,对苯并咪唑中N原子的位置进行转移,改为咪唑并吡啶结构可以调整整个分子的平面性,所得化合物49的pKa增加, cLogP降低,从而提高水溶性,ADME特性也有所改善。
06苯并萘啶酮类及喹啉类
首先,针对mTORC1的中通量筛选获得了先导化合物50,后引入六元内酰胺形成新的三环骨架并进行结构修饰得到Torin1(51),在纳摩尔浓度下抑制mTORC1和mTORC2的磷酸化,Torin1对mTOR的选择性比PI3K高900倍。但其较差的稳定性和较低的口服生物利用度限制了进一步的体内研究。对Torin1进行改造,移除代谢位点基团,减少分子量,进一步提高水溶性,得到Torin2(52),稳定性提高。对Torin 2与mTOR结合模式进行分析,α,β-不饱和内酰胺结构十分重要,它与喹啉环的C-3和C-4位稠合限制了间三氟甲基苯环的构象,然而,作为迈克尔受体,α,β-不饱和内酰胺很容易在体内被捕获,从而导致Torin 2的清除半衰期较短。将六元内酰胺环开环,并引入分子内氢键以模拟其生物活性构象,设计得到的喹啉衍生物53是有效的mTOR抑制剂,IC50为14nM,细胞活性也很强。

图:苯并萘啶酮类及喹啉类mTOR抑制剂
NSC781406(54)是一种新型有效的PI3K/mTOR双重抑制剂,在60种癌细胞中的平均GI50为65nM,其中4种癌细胞系的GI50小于10nM,此外,在肝癌细胞异种移植模型中也表现出有效的肿瘤生长抑制作用,效果优于索拉菲尼。GSK1059615(56)是一个进入临床的PI3K抑制剂,共晶结构表明噻唑烷二酮与PI3Kγ的ATP结合口袋内的催化赖氨酸(Lys833)存在相互作用,但该空腔还可以容纳一个更大的基团,基于GSK1059615的改造发现了GSK2126458(55),是一种mTOR/PI3K双重抑制剂,对mTORC1的Ki值为0.18nM,对mTORC2的Ki值为0.3nM。GSK2126458具有较低的血浆清除率和良好的口服生物利用度,体内实验也表现出优异的活性和良好的耐受性。
07咪唑并喹啉酮类
咪唑并[4,5-c]喹啉骨架能够模拟ATP的腺嘌呤部分以多种结合模式与激酶的铰链区产生氢键相互作用。NVP-BEZ235(57)是由诺华开发的一种口服mTOR/PI3K双重抑制剂,对mTOR的IC50为20.7nM,对PI3Kα, β, δ 和γ的IC50值为4nM、75nM、7nM和5nM。NVP-BGT226(58)是诺华开发的另一个口服mTOR/PI3K双重抑制剂,具有一定的细胞毒活性,在肝癌,头颈癌,胰腺癌,非小细胞肺癌中均进行了研究。LY3023414(59)是礼来公司开发的一种口服mTOR/PI3K双重抑制剂,对mTOR的IC50为165nM,对PI3Kα、β、δ和γ的IC50值分别为6.07nM、77.6nM、38nM和23.8nM。与其他PI3K/mTOR抑制剂相比,该化合物的溶解度和溶出特性良好,有利于吸收,所以具有高生物利用度。
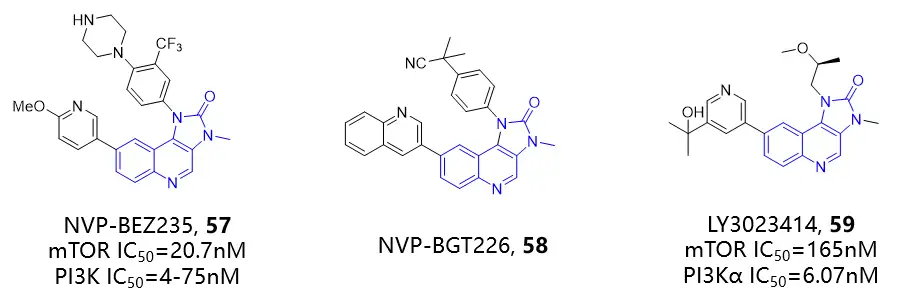
图:咪唑并喹啉酮类mTOR抑制剂
08其他母核
PI-103(60)是第一个公认的mTOR/PI3K双重抑制剂,是日本制药公司Astella通过高通量筛选及结构优化得到。对mTOR的IC50为20nM,对PI3K的IC50为3nM。PI-103对一些肿瘤细胞有很好的抑制作用,但由于成药性差没有进入临床。针对mTOR活性的高通量筛选得到一个含有新骨架的抑制剂61,进一步的优化得到该系列活性最好的化合物XL388(62),具有中等的生物利用度,良好的药代动力学和口服暴露。PKI-402(63)具有三唑并嘧啶的母核结构,其对mTOR、PI3Kα的IC50值分别为1.7nM、1.4nM,对MDA-361细胞异种移植肿瘤的体内药效研究表明,以100mg/kg剂量iv给药导致肿瘤消退,70天内肿瘤没有再次生长。

图:其他母核的mTOR抑制剂
PF-04691502(64)能有效抑制PI3K/mTOR信号通路,抑制肿瘤细胞增殖,在SKOV3卵巢癌异种移植模型和4个NSCLC异种移植模型(NCI-H460、A549、NCI-H1650和NCI-H1975)中显示出强大的抗肿瘤活性。VS-5584(65)对细胞PI3K/mTOR通路有着显著调节作用,抑制PI3K和mTORC1/2下游底物的磷酸化,在广谱癌细胞中显示出高抗增殖活性,对许多雷帕霉素抗性细胞系有效。GDC-0084(66)是一种有效的基于嘌呤结构的PI3K/mTOR抑制剂,它对mTOR的Ki值为70nM,对PI3Kα的Ki值为2nM,能够穿透血脑屏障,在U87异种移植模型中抑制肿瘤生长,且耐受性良好。OSI-027(67)对mTOR的IC50为4nM,在不同癌细胞系(BT-474、IGR-OV1和MDA-MB-231)中有效抑制mTORC1和mTORC2,具有广谱抗肿瘤活性,在乳腺癌、结肠癌、肺癌、前列腺癌、淋巴瘤和头颈癌等几种模型中显示出显著的肿瘤生长抑制,且耐受性良好,在治疗期间观察到的体重减轻不足10%。

图:其他母核的mTOR抑制剂
高通量筛选获得的1,6-取代的咪唑并[4,5-b]吡嗪-2-酮化合物68,mTOR抑制IC50为1.27μM。进一步的构效关系优化得到化合物69,mTOR抑制IC50为2nM,在PC3肿瘤细胞中实现了mTOR通路抑制,阻断了mTORC1和mTORC2的信号传导。CC214-1(70)与化合物69相比与四氢吡喃环连接的链长增加,对mTOR也具有很好的抑制活性。通过亚甲基单元的插入进行扩环、更改母核等策略设计了一系列新的化合物,这些类似物保留了对PI3Kα的选择性,但对mTOR的效力下降。CC214-2(71)是其中具有代表性的化合物,以剂量依赖的方式显著抑制PC3细胞生长。进一步的构效关系研究旨在提高对mTOR的活性,改善口服药代动力学,得到了CC-223(72),它具有优异的理化性质和药代动力学特性,出色的激酶选择性,在多种口服给药的体内实体瘤模型中证明了疗效,此外还有良好的体内和体外安全性。XL-765(73)由单靶点PI3K抑制剂XL-147(74)修饰而得到,实验数据尚未发表,它已进入I期临床试验,包括单次给药和与其他药物联合,如替莫唑胺和厄洛替尼,是一种有希望的ATP竞争性双重PI3K/mTOR抑制剂。
第三代mTOR抑制剂
第三代mTOR抑制剂RapaLink-1(75)是将雷帕霉素和mTOR激酶抑制剂MLN018整合到同一个分子上而获得的,其通过结合FKBP12的FRB结构域作用于mTOR,同时连接的小分子抑制剂MLN018作用于mTOR激酶结构域。

图:RapaLink-1的结构
天然产物类mTOR抑制剂
除了上述介绍的第一代、第二代、第三代mTOR抑制剂以外,一些天然产物也被报道能直接或间接抑制mTOR和mTOR信号通路,如姜黄素(Curcumin, 76),EGCG((−)-Epigallocatechin gallate, 77),白藜芦醇(Resveratrol, 78),萝卜硫素(Sulforaphane, 79)等。

图:天然产物类mTOR抑制剂
总结
mTOR是一种关键的信号转导分子,位于mTOR信号通路的中心,参与调节细胞生长、增殖、存活和自噬等过程。mTOR信号通路障碍与癌症、糖尿病、阿尔茨海默病和自身免疫性疾病等密切相关。尽管变构mTOR抑制剂的发现较早,对Rapalogs的研究已经成熟,但Rapalogs分子量大,手性碳多,合成难度较大,此外,位点修饰有限。因此,该类mTOR抑制剂未来的发展方向将是在现有药物的基础上做一些简单的改造。
靶向ATP结合位点的小分子mTOR抑制剂的合成相对容易,对mTOR的抑制更为彻底。因此,小分子mTOR抑制剂也将是未来的一大研究方向。由于第二代mTOR抑制剂的耐药机制,第三代mTOR抑制剂的开发一直备受药理学家和药物化学家的关注。第三代mTOR抑制剂通过Linker将雷帕霉素与ATP竞争性mTOR抑制剂连接起来,从而抑制耐药突变体,是重要的研究突破。总而言之,mTOR抑制剂的开发和探索之路还需要各界研究人员的共同努力。
参考文献:
1.Yifan Chen, Xiaoping Zhou. Researchprogress of mTOR inhibitors.Eur.J. Med. Chem.2020, 208, 112820.
2.Tian Xua, Dejuan Sun, et al. Targeting mTORfor fighting diseases: A revisited review of mTOR inhibitors.Eur. J. Med. Chem.2020, 199, 112391.
本文版权归原作者所有,文章内容不代表平台观点或立场。如有关于文章内容、版权或其他问题请与我方联系,我方将在核实情况后对相关内容做删除或保留处理!联系邮箱: yzhao@koushare.com