引言
讲到药物发现的小故事,就不得不提教科书上的典型案例西咪替丁。众所周知,西咪替丁是一种治疗溃疡的H2受体拮抗剂。这里的溃疡指的可不是“溃疡不是病,疼起来要命”的口腔溃疡,而是一种多发的常见病消化性溃疡,主要发生于胃和十二指肠中。不规律不健康的饮食习惯,情绪波动过大,吸烟喝酒,幽门螺杆菌,遗传因素,甚至是一些药物都可能引发胃溃疡。从生理角度上来讲,“无酸无溃疡”,胃酸分泌过多导致胃黏膜缺损,是引起溃疡的直接因素。
要治疗一种疾病就得对症下药,胃酸分泌过多是导致溃疡发生的重要原因,那第一个想到的治疗方法就是把多余的胃酸中和掉,所以出现了【抗酸剂】这类药物,比如碳酸氢钠、氢氧化铝。但这也只是治标不治本,大家就想,能不能从根本上来抑制胃酸的分泌。首先我们得搞清楚胃酸是怎么分泌的。在人体的胃壁细胞上存在着很多受体,其中与胃酸分泌比较相关的就是组胺(Histamine),乙酰胆碱(Ach)和胃泌素(Gastrin)受体。“一把钥匙开一把锁”,组胺,乙酰胆碱,胃泌素这些信号分子可以激活各自的受体,并产生一系列的刺激,这些刺激经过第二信使cAMP和Ca2+的作用,启动质子泵(H+,K+-ATP酶),将氢离子泵向胃腔,氢离子与顶膜转运至胃腔的氯离子结合,就形成了形成胃酸的主要成分——盐酸。

图: 胃酸分泌的机制
根据这个过程,有两个地方可以做文章,一是阻断相应的受体,也就是开发【组胺受体抑制剂】,【乙酰胆碱受体抑制剂】,【胃泌素受体抑制剂】;二是阻断质子泵,开发出【质子泵抑制剂】。今天的主角西咪替丁就是通过阻断组胺受体来起到抑制胃酸的作用。
当然这些理论都还是后来补充发现的。在没有弄清楚这些机制之前,西咪替丁又是怎么被开发出来的呢?
大胆假设:组胺受体有不同亚型?
20世纪50年代,人们就已经发现,组胺可以刺激胃酸的分泌。当时已经发现了一些抗组胺药物,比如苯海拉明可有效地减弱组胺的许多反应,用于抗过敏疾病。但令人疑惑的是,这个抗组胺药物并不能减少胃酸分泌。既然出现了与实际情况不符合的问题,就总有好奇的研究人员去探究背后的原因。1963年,英国帝国化学工业集团(ICI集团)的James Black提出假说,认为胃部存在着不同亚型的组胺受体(后来我们称之为H2受体),很可能是因为组胺与这种亚型的受体结合,才导致了胃酸的大量分泌。

图:组胺受体可能存在不同亚型
可惜的是,他的公司对他的这个猜想不以为意,觉得琢磨一个机制这么复杂而且还没搞清楚的东西是浪费时间浪费金钱。得不到支持的James Black毅然决然跳槽去了另一个公司Smith Kline &French公司(也就是后来的史克公司),踏上了探索组胺受体亚型和寻找组胺受体拮抗剂的漫漫长路。
万事开头难:4-甲基组胺,一个走向相反方向的分子
由于之前并没有相关的抑制剂被报道,也不清楚组胺受体的具体结构和作用模式,Black只能先从组胺分子的结构改造出发。新药物的发现总是困难重重,一晃就四年过去了,研究小组合成了200多个组胺的衍生物,然而并没有得到有预期效果的分子。
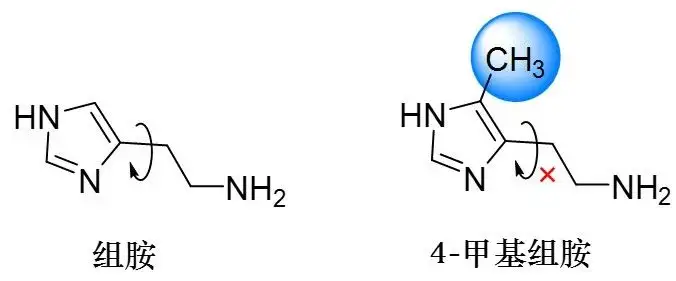
图: 组胺和4-甲基组胺的结构式
Black觉得这么瞎找盲筛不可行,得从经验教训中发现一点规律。之前的研究发现了4-甲基组胺,可惜是一种能促进胃酸分泌的H2受体激动剂,激动作用还比组胺更强。这似乎和原来的研究方向背道而驰了,但从这个分子身上Black看到了一点启发。他猜想是由于甲基增加了位阻,限制了侧链的自由旋转。于是他们决定暂时保留组胺的咪唑部分,而对氨基侧链进行改造。
稍有眉目:N-胍基组胺,部分拮抗作用
他们对组胺的存在形式进行了分析,发现在胃的生理pH(酸性)条件下,组胺分子是以正离子(NH3+)的形式存在,侧链的电子云密度有所下降。这个关键信息提示他们要寻找H2受体的拮抗剂,说不定可以从降低侧链氮原子的电子密度着手。于是他们结合了物理化学药学等方面的知识,改造发现了N-胍基组胺,这是一个组胺受体的激动/拮抗剂。这个分子的出现又给了他们一点提示,他们猜想H2受体存在两个阴离子性质的部位,就像弹簧开关一样,H2受体上存在三个弹簧,咪唑的氮可以触发第一个,组胺的正电性的氨基可以触发第二个,当第一个和第二个弹簧都被触发的时候,就可以激活这个受体;但只有当第三个弹簧也被触发,才能达到拮抗的效果。所以氨基后面应该还需要加上一个额外的功能基团。
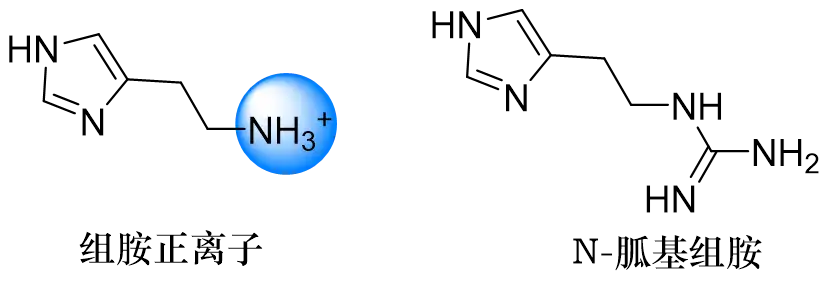
图: 组胺正离子和N-胍基组胺的结构式

图: 假想的H2受体拮抗剂作用方式
改造改造再改造:SKF91486和SKF91581
根据这个思路,增加N-胍基组胺的链长,得到了化合物SKF 91486,但这个化合物仍有部分激动作用。他们又想到运用药物设计中十分常见又经典的电子等排原理,将胍改为硫脲,并且把硫脲基团甲基化,得到SKF 91581。他们发现侧链氨基与受体的激动剂键合部位结合,末端氨基与受体的拮抗剂键合部位结合,证明了上面的“弹簧理论”。此外,令他们惊喜的是,将胍改为硫脲衍生物这一操作,降低了碱性的同时也提高了拮抗活性。
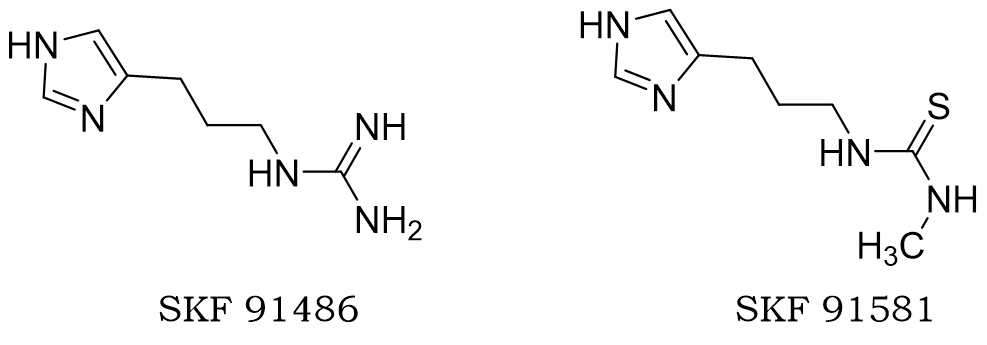
图: SKF 91486和SKF 91581的结构式
胜利曙光:布立马胺,终于没有了激动活性;甲硫米特,活性进一步提高!
结合两种策略,既增加链长又降低碱性,在1971年得到了布立马胺(Burimamide),它是研究团队得到的第一个H2受体的竞争性拮抗剂,无激动作用,胜利的曙光仿佛就在眼前了,这时距离项目开始已经过去了七年。
但是,也不能高兴得太早,布立马胺存在一个较大的问题,就是口服活性太差。为了提高口服活性还得进行进一步改造。
怎么提高布立马胺的活性呢?还得从仔细分析组胺分子着手。组胺是一种咪唑衍生物,存在[1,4]和[1,5]两种互变异构体。咪唑正离子,[1,4]-互变异构体和[1,5]-互变异构体这三种存在形式之间存在质点平衡。布立马胺的主要质点之一是正离子(40%),[1,4]-互变异构体最少;而组胺的主要质点是[1,4]- 互变异构体(近80%),正离子只占少部分(约3%)。两者占优势的质点各不相同。那是不是这两者在存在形式方面的差异导致布立马胺口服活性低呢?
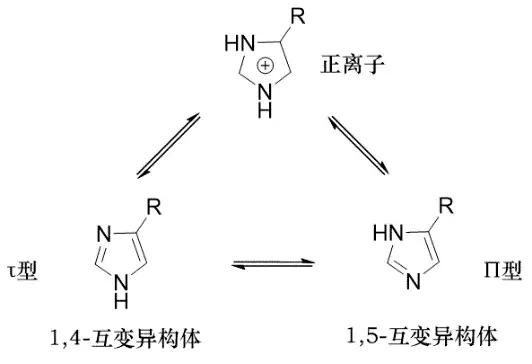
图: 咪唑衍生物的质点平衡
他们认为,如果拮抗剂的活性质点主要是与组胺相同的[1,4]-互变异构体,则拮抗作用可能增强。他们又发现,互变异构体的量和R基有很大的关系,当R基为给电子基时,[1,5]-互变异构体的量增加,反之当R为吸电子基,[1,4]-互变异构体较多。组胺的侧链,因电负性较大的β-氨基的作用传递到咪唑环,具有吸电子效应;而布立马胺的侧链为δ-取代烷基,给电子效应。所以要进一步改造,使布立马胺侧链呈现与组胺侧链相同的吸电子性。

图: 布立马胺、硫代布立马胺和甲硫米特的结构式
不管干啥,图方便总是大家的共性。他们考虑到合成方面的方便性,将布立马胺侧链上的第二个亚甲基换成电负性较强的电子等排体-S-,得到硫代布立马胺,并且一举成功,证实它的主要存在形式是[1,4]-互变异构体了,对H2受体的拮抗作用比布立马胺要高3倍。但将亚甲基改为-O-,效果就没那么好。
再结合之前说到的4-甲基组胺,他们又进行了进一步的分析,觉得4-位甲基是一个给电子基,也可能会影响邻位的氮原子,于是试着在硫代布立马胺的4位也引入一个甲基,得到甲硫米特(Metiamide)。甲硫米特的拮抗活性比布立马胺强8-9倍,是第一个进入临床试验的H2受体拮抗剂,疗效明显治愈率高。
手牵手过河:甲硫米特遭到禁用
然而,令人痛心的是,由于在一些病人身上观察到肾损伤和粒细胞缺乏症,甲硫米特的临床试验被迫终止了。听到这个消息,Black心态都要崩了,“我们接到公司的电话, 说甲硫米特遭到禁用。之后,我们不得不牵着手过河,因为深怕有人会跳河,每个人都沮丧得不得了。”但是他并没有放弃,还是选择硬着头皮做下去。
千难万险:西咪替丁问世
他们推测可能是侧链上硫脲的硫原子造成了这种毒副作用,于是用硫脲的电子等排体进行了一系列的替换和构效关系研究,最终采用了氰基胍替换的化合物西咪替丁(Cimetidine)进行临床研究,西咪替丁疗效优于甲硫米特,且没有粒细胞减少的副反应,成为第一个上市(1977年,英国)的H2受体拮抗剂,商品名为Tagamet®(中文:泰胃美),长达十几年的开发历程暂时画下了一个圆满的句号。
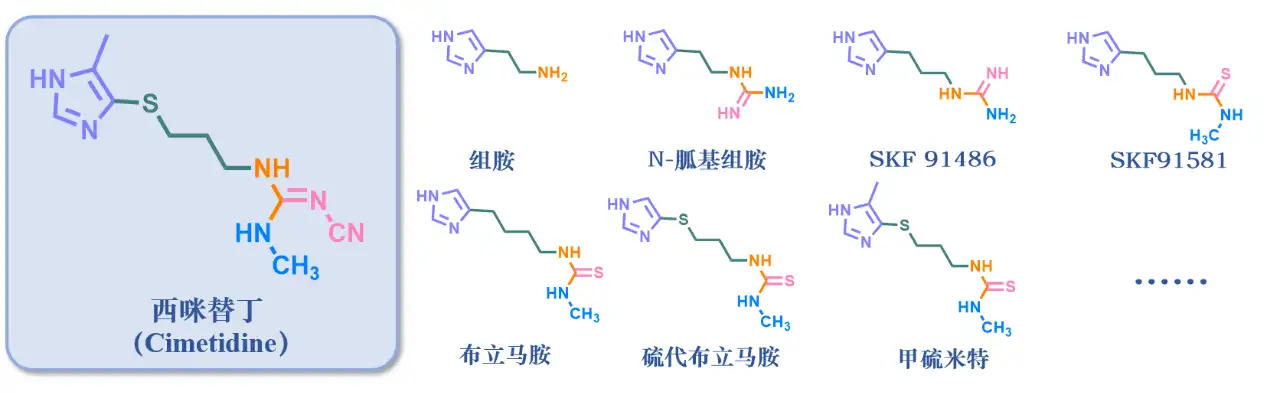
图: 西咪替丁的发现历程
西咪替丁是合理药物设计的典范,开发历程长达十几年。西咪替丁一经问世即成为治疗溃疡的首选药物,刚上市时的价格是20美元/100粒,是第一个年销售额超过10亿美元的药物,之后在世界上100多个国家获准上市,被称为“重磅炸弹”。西咪替丁项目的负责人Black教授也因此获得1988年诺贝尔奖。
在西咪替丁之后,出现了呋喃类的第二代H2受体拮抗剂,如雷尼替丁(Ranitidine),打破了咪唑环是H2受体拮抗剂的必要结构的设定,第三代噻唑类如法莫替丁(Famotidine)药效更强。关于H2受体拮抗剂的研究仍未完待续。
原文刊载于【Hello药学】公众号
本文版权归原作者所有,文章内容不代表平台观点或立场。如有关于文章内容、版权或其他问题请与我方联系,我方将在核实情况后对相关内容做删除或保留处理!